

To Issue 145
Citation: Parry M, “Considerations when Switching from Current pMDI Propellants to New Lower-GWP Propellants”. ONdrugDelivery, Issue 145 (Apr 2023), pp 6–9.
Mark Parry discusses the considerations that need to be made when switching from current pressurised metered dose inhaler propellants to new alternatives with lower global warming potential, including formulation, regulatory and analytical standpoints.
INTRODUCTION
As climate change continues to drive political and legislative action, many organisations are striving to achieve net zero greenhouse gas emissions. For manufacturers of inhaled medicines, this means exploring more sustainable inhaler propellants. Switching from current pressurised metered dose inhaler (pMDI) propellants to new propellants with lower global warming potential (GWP) could help to reduce the carbon footprint of pMDI-delivered medicines significantly.
“pMDIs represent a significant portion of carbon emissions in the medical sector, with an estimated 3.1% of emissions being due to these products, according to the UK NHS.”
The Montreal Protocols1 in the 1990s drove the migration from chlorofluorocarbons (CFCs) to hydrofluorocarbons (HFCs) to address the critical need to move away from ozone-depleting chemicals – but also started a move to lower-GWP fluorinated-gas (F-gas) propellants. Evolution of these regulations with the Kigali amendment1 aims to further reduce the use of higher-GWP F-gas propellants and promote the development and adoption of new low-GWP propellants. The structure of these targets is phased over time and across different regions but targets an 80% reduction in the use of current pMDI propellants in the EU by 2030, providing a strong incentive to migrate to these newer low-GWP options.
DEVELOPMENT APPROACH
Addressing the Carbon Footprint pMDIs represent a significant portion of carbon emissions in the medical sector, with an estimated 3.1% of emissions being due to these products, according to the UK NHS.2 Migration to alternative device technologies, such as dry powder inhalers (DPIs), has been suggested as a route to mitigate these risks. However, it should also be understood that the vast majority of emissions from pMDI devices are due to the propellant and reformulation to low-GWP propellants could potentially close, or even beat, the gap between pMDIs and their DPI and soft mist inhaler (SMI) alternatives.2
Understanding the overall carbon footprint of an inhaler is also more complex than just the choice of propellent gas, and companies looking to address their corporate social and environmental responsibilities need to look at the whole product lifecycle. This includes considering the production and transport of all the constituent device and formulation components, as well as distribution and disposal of the finished product. For example, inhalers make significant use of virgin-plastic materials and lack widescale recycling support. Some companies are making efforts to address this, such as Boehringer Ingelheim’s development of a reusable version of its Respimat device platform, and it is important to look at the full context and complete lifecycle of a product.
Propellent Availability
The use of F-gases for pMDI products represents around 2.3% of F-gas contributions to greenhouse gas emissions,3 with the use of F-gases as a refrigerant by far the most dominant use. This could suggest that continued use of these gases for medical purposes may be supported within the global targets; however, the availability of high-purity, pharma-grade propellants at reasonable prices is a result of the large-scale production of these gases to support their use as a refrigerant. As production volumes reduce, the costs and availability of pharma-grade gases will increase significantly and so, regardless of a company’s broader carbon-reduction strategies, addressing the migration away from current HFA propellants will have both regulatory and commercial drivers.
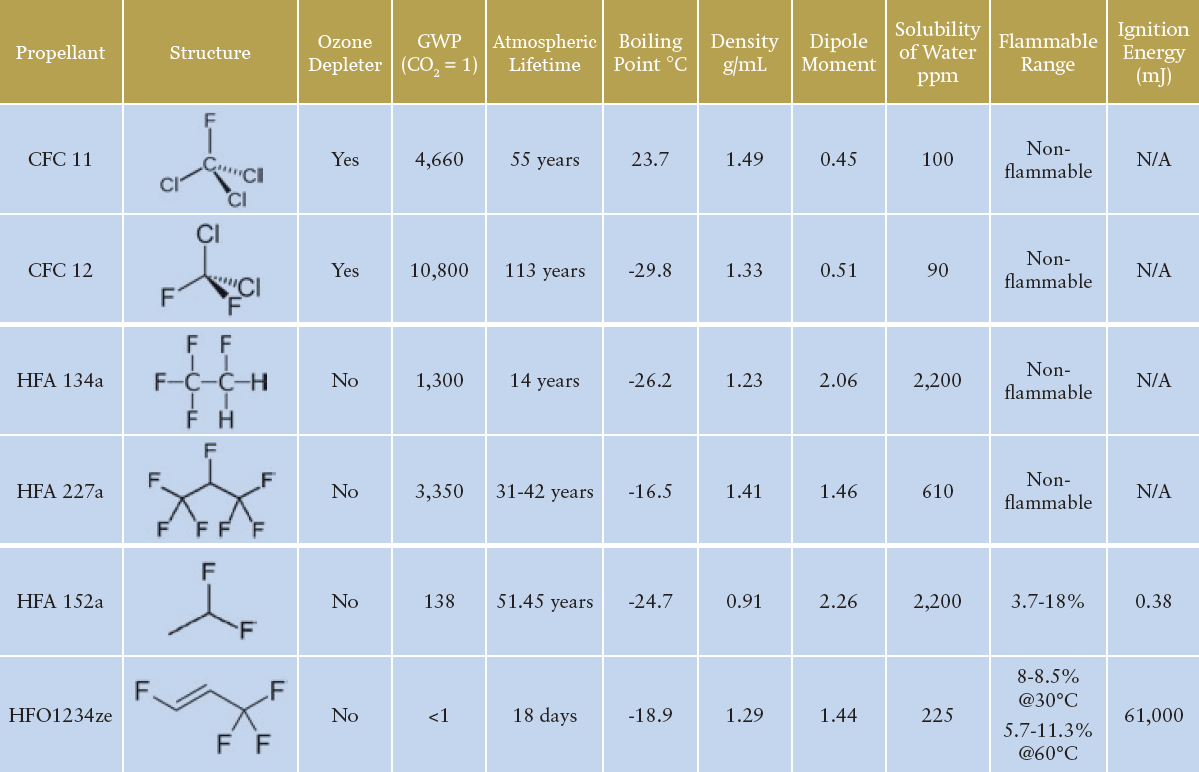
Table 1: An overview of the primary historic, current and future propellants of interest for pMDI formulations.3,4
“Whichever propellent is selected, despite the somewhat similar physical properties, reformulation of existing products may not be as simple as a drop-in replacement of the new propellent.”
Propellent Choice
Table 1 illustrates the key propellants under discussion. HFA 134a and HFA 227a are the propellants currently in widespread use for pMDI products, with HFA 134a representing the major proportion of current usage. HFA 152a and HFO1234ze are the primary new low-GWP propellant options being used in the development of new and reformulated pMDI products. Both new options provide physical properties that suggest some compatibility with the current HFA propellants – indeed, both are being used successfully by companies such as Chiesi and AstraZeneca. The question of flammability, however, should be addressed.
HFA 152a is classified as an extremely flammable gas, with a minimum ignition energy within the range of a typical static discharge from a human. The concentration range needed for flammability does mean the risk is likely not significant for a patient in normal use, but it does present new challenges for identifying, assessing and managing the risk involved in handling the raw material and manufacturing the drug product. HFO1234ze is technically classified as non-flammable, however, it does have a flammability range at elevated temperatures and is generally classified as mildly flammable in its widespread use as a refrigerant.
While the incorporation of ethanol in existing HFA 134a-based formulations has presented some flammability challenges for current pMDI products, the particular challenges of HFA 152a may mean that significant investment in redesigning or upgrading manufacturing facilities is required to ensure that it can be handled safely.
Formulation
Whichever propellent is selected, despite the somewhat similar physical properties, reformulation of existing products may not be as simple as a drop-in replacement of the new propellent. The general principles of pMDI formulation still apply, with solution and suspension formulations achievable and currently used excipients still applicable if required.
Considering suspension formulations, it may be possible to maintain the engineered API particle size across a change in propellant. However, the stability and performance of the suspension formed will depend greatly on the physicochemical properties of the propellent, such as its density and how it interacts with the surfaces of the micronised API. Examining the sedimentation stability of salbutamol sulphate suspensions in HFA 134a, HFA 152a and HFO 1234ze showed sedimentation rates of 188 ±26, 226 ±136 and 60 ±24 mm/hr respectively, while HFA 227a showed creaming behaviour with the API not fully settling, likely due to the propellant’s higher density.5
Looking at solution formulations, the produced particle size from the device will depend more significantly on the characteristics of the aerosol spray, such as droplet size. Factors like the evaporation rate, solubility and density will all have an effect and research has shown differences in droplet size and spray stability when looking at both placebo and model formulations.6 The change in propellent can also influence the API solubility and may require changes in the co-solvent levels if migrating a solution formulation. The work referenced looked also at salbutamol sulphate and beclomethasone dipropionate solubility in HFA 134a, HFA 227a, HFA 152a, and HFO 1234ze.5 Salbutamol showed no solubility in any of the propellants, while the solubility of beclomethasone dipropionate in HFA 152a was 400 µg/mL – more than twice the concentration observed with HFA 227a and three times the concentration observed with HFA 134a and HFO 1234ze.
When considering new chemical entities (NCEs), incorporating these new propellants into formulation development should be reasonably straightforward – provided that the development facilities address the increased handling risks. For reformulation projects, neither of the new propellants provide complete equivalence with those currently in use, and so a broader formulation development design should be considered to understand the specific behaviours and interactions between the API and the propellants.
“Engaging early with the relevant regulatory parties and understanding the likely scope of the changes that are needed for a given product will be a key factor in ensuring that the development process is as focused, pragmatic and efficient as possible.”
REGULATORY APPROACHES
For NCE products, incorporating these new propellants into product development should be more straightforward through the relevant NDA and marketing authorisation application (MMA) routes. Some work regarding the toxicology and safety of the new propellants will need to be incorporated, and the manufacturers of these propellants are working to support these studies and establish the drug master files to support their integration.7
Reformulating Products
When considering the reformulation of existing products, the regulatory approach is more complex. There is no specific guidance directing the industry on how to approach these reformulation projects; however, two main approaches present themselves – either treating it as a generic or as an NDA.
Approaching as a “hybrid” generic product via a US FDA Accelerated NDA 505(j) application seems logical, as the objective is likely to replicate the current product with the new propellent. This may prove appropriate, however, modern expectations for bioequivalence have continued to develop and expand since some of the original products were developed.8
The successful approval of inhaled generic products continues to prove challenging for the industry with only a handful of generic pMDI products on the market, highlighting the difficulty of demonstrating equivalence even without a change in one of the key formulation elements. With the major formulation component – the propellant – changing to a new material, the generic-like approach is potentially a significant challenge, due to the evolving toxicological and safety positions for these new propellants, the likely need to change other elements of the formulation to provide clinical equivalence and the likely change in spray characteristics that may impact in vitro equivalence.
Approaching the product as an NDA is likely to be the more prevalent approach for these products, even though the API has an established safety and clinical data profile. Looking back to the CFC-to-HFA migration that started in the 1990s, this was generally the approach taken for those products. This provides the space to address more significant changes in formulation, while still using data from the existing approved product; however, it does mean a significant increase in expected costs and timelines overall.
The ecological urgency that drove migration away from CFC use is less prominent here and, even then, full replacement of the CFC pMDI market in the US took until 2014 to complete9 – 25 years after the initial experiments with HFA-based formulations and 18 years after the 1996 target in the Montreal Protocol for the end of all non-exempted use. The regulatory position surrounding these reformulation projects is still evolving but, given the targets of the latest amendments to the F-gas regulations, it is likely regulators will continue to take a measured, but conservative, approach to reviewing these products. Engaging early with the relevant regulatory parties and understanding the likely scope of the changes that are needed for a given product will be a key factor in ensuring that the development process is as focused, pragmatic and efficient as possible.
ANALYTICAL CONSIDERATIONS
When looking at the reformulation of an existing product, there is also a need to review the analytical methodology and overall chemistry, manufacturing and controls approach. Even if there are no significant changes in the analytical strategy, improvements in technology may yield improvements in method reliability, consumable use and overall turnaround time that can have a significant positive impact on the costs of the analytical activity.
With changes being made to the formulation, a suitable validation gap analysis of methods will be required, even if no changes are made to the procedures. These methods will underpin the generation of significant volumes of data, such as product characterisation, stability and in vitro bioequivalence work, so any validation should be addressed early in the process. Depending on the development and regulatory approach taken, it may be possible to use data from the existing product; however, care should be taken to ensure these data are evaluated against the current regulatory expectations.
Generics and In Vitro Bioequivalence
Whether or not a generic approach to the regulatory pathway is taken, understanding the current expectations and techniques used for building a strong understanding of in vitro bioequivalence is valuable. Current regulations still have an understanding of emitted dose and aerodynamic particle size at their core, but also include expectations for equivalence in priming and re-priming behaviour, spray pattern and plume geometry, which may prove more challenging to meet with a change in propellant.
Beyond the core regulatory expectations, considerable work has been undertaken to expand the range of analytical tests addressing in vitro-in vivo correlation, with the aim of both de-risking and reducing the need for clinical studies for generic product developments. Even if the generic approach isn’t being followed, these can provide additional insight into product performance during development and how the product might perform in the clinical setting. For pMDIs in particular, the use of spray pattern/plume geometry testing,10 as well as plume velocity and actuation force measurements,11 can generate key insights into aerosol and device performance.
Other developments, such as the incorporation of physiologically relevant throat models and breathing profiles, as well as the use of morphologically directed Raman spectroscopy,12 have provided key advances – in particular for DPI products – that may also provide useful insights here.
CONCLUSION
There is a combination of environmental, regulatory, commercial and ethical drivers pushing the industry towards adopting lower-GWP propellants for pMDI products as part of the overall management of their carbon footprint. The availability of new propellants, such as HFA 152a and HFO 1234ze, is scaling up with new manufacturing plants for both coming online in the last year. Both are well positioned for broad market adoption, although the handling risks of HFA 152a in particular will need to be addressed to incorporate these into existing manufacturing facilities safely.
Work looking to formulate products using these new propellants has shown them to be viable replacements, but also highlights that there will be development challenges to address in migrating existing products. The regulatory position is still developing for these reformulation projects, and collaboration and consultation between regulators and businesses will be important in developing best practices and expectations in this area, and ensure the commercial feasibility of an important legislative drive to tackle climate change.
REFERENCES
- Harwick M, “Evolving Legislation Associated with Fluorinated Hydrocarbon Propellants used in Metered Dose Inhalers”. Resp Drug Deliv 2022, 2022, Vol 1, pp 55–64.
- Fulford B et al, “Carbon Footprints and Life Cycle Assessments of Inhalers: A Review of Published Evidence”. Sustain, 2022, Vol 14(12), article 7106.
- Pritchard JN, “The Climate is Changing for Metered-Dose Inhalers and Action is Needed”. Drug Des Devel Ther, 2020, Vol 14, pp 3043–3055.
- Atkinson N “Considerations for Robust Clinical and Commercial Manufacturing of Next Generation Sustainable Metered Dose Inhalers”. Resp Drug Deliv, 2022, Vol 1, pp 75–86.
- Shur J et al, “The Formulator’s Guide to Transitioning to Low Global Warming Potential pMDIs”. Resp Drug Deliv, 2022 Vol 1, pp 65–74.
- Rao L, “In-Vitro Evaluation of pMDI Spray Development of HFA134a, HFA152a and HFO1234ze(E)”. Conference Poster, Drug Delivery to the Lungs (DDL), Edinburgh, UK, Dec 7–9, 2022.
- Kuehl PJ, Corr S, Leach CL, “Safety, Tolerance and Pharmacokinetics of HFA-152a in Healthy Volunteers”. Resp Drug Dev, 2022, Vol 1, pp 87–96.
- Lee SL et al, “Regulatory Considerations for Approval of Generic Inhalation Drug Products in the US, EU, Brazil, China, and India”. AAPS J, 2015, Vol 17(5), pp 1285–1304.
- “Market Characterization of the U.S. Metered Dose Inhaler Industry”. ICF, Sep 2021.
- Jeanneret M et al, “Candidate Device Selection for pMDI In-Vitro only Bioequivalence focussing on Spray Pattern and Plume Geometry Analysis”. Conference Poster, Drug Delivery to the Lungs (DDL), Edinburgh, UK, Dec 7–9, 2022.
- Houlden J et al, “Plume Front Velocity and Force to Actuation Characterisation of Pressurised Metered Dose Inhalers and Soft Mist Inhalers”. Conference Poster, Drug Delivery to the Lungs (DDL), Edinburgh, UK, Dec 8–10, 2021.
- Davies D, “Investigation of DPI Particle Microstructure by MDRS to Explain Differences Observed in NGI Results Between Equivalent Products”. Conference Poster, Drug Delivery to the Lungs (DDL), Edinburgh, UK, Dec 11–13, 2019.

