To Issue 130
Citation: del Amo EM, “Pharmacokinetic Models: Indispensable Tools for Ophthalmic Drug Development”. ONdrugDelivery, Issue 130 (Mar 2022), pp 59–63.
Eva Maria del Amo presents an overview of pharmacokinetic models for ophthalmic drugs, and discusses the value of these models for future drug development in the sector.
Financing in the global ophthalmic pharmaceutical sector has increased eightfold over the last 20 years, with an estimated value of US$42.1 billion (£31.4 billion) in investment by 2024.1 Ophthalmic diseases can affect either the anterior segment of the eye, such as dry eye, allergy, infection, glaucoma and inflammation (e.g. conjunctivitis); or the posterior segment, such as age-related macular degeneration, diabetic retinopathy, retinal vein occlusion and neural changes induced by glaucoma. The prevalence of the posterior-affecting diseases is increasing in line with ageing populations, making retinal treatments a subject of particular interest. Such treatments are now being actively investigated in the pharmaceutical industry and academia, with a focus on new strategies to prolong and improve drug delivery to the retina.
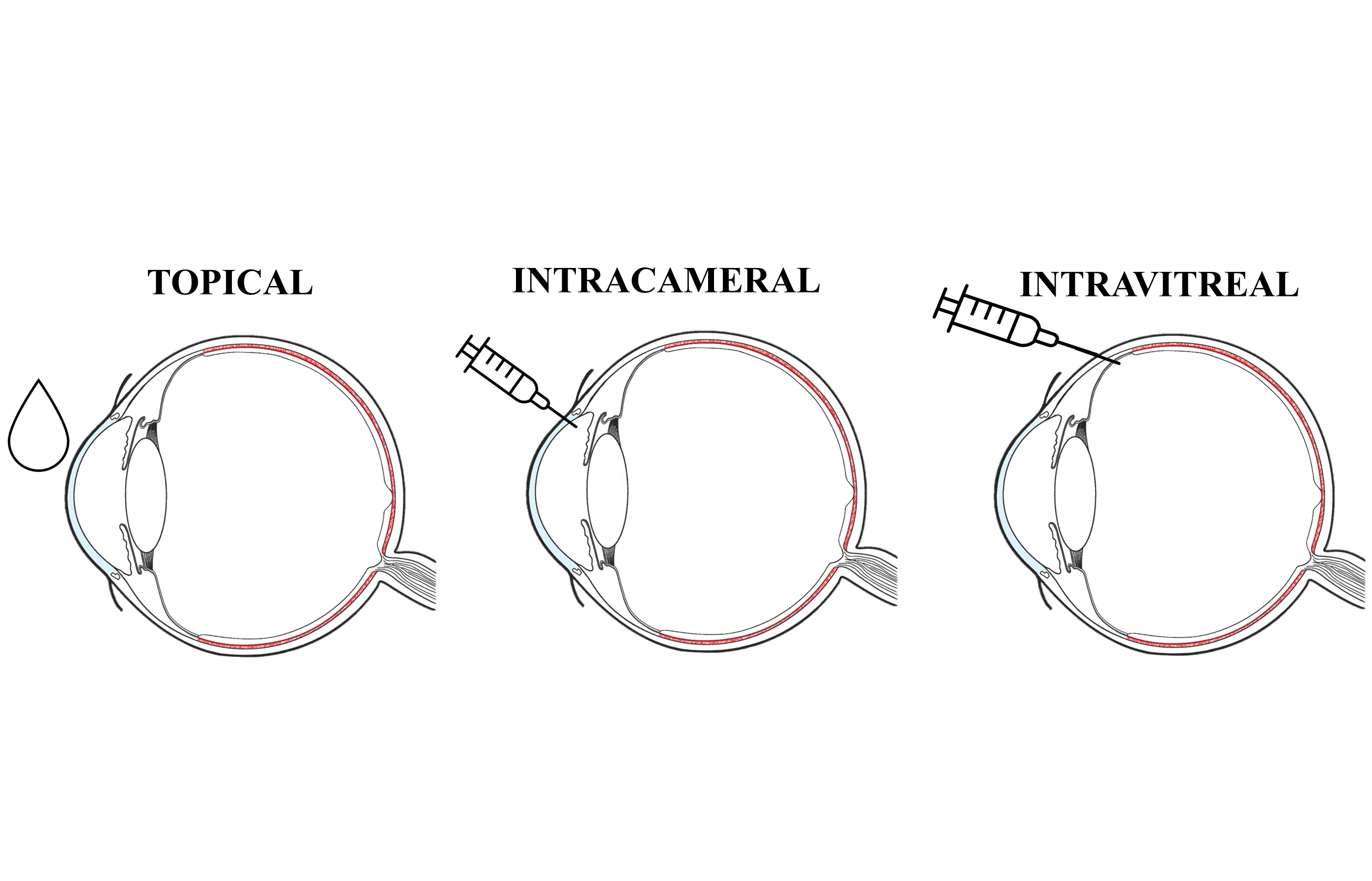
Understanding the physiology of the eye and the relevant barriers to each route of administration and each type of drug (low molecular weight drugs (small drugs), biologics or controlled-delivery systems) is essential. The topical and intravitreal administration routes currently represent the gold standard for treatment of the anterior and posterior segments respectively (Figure 1).
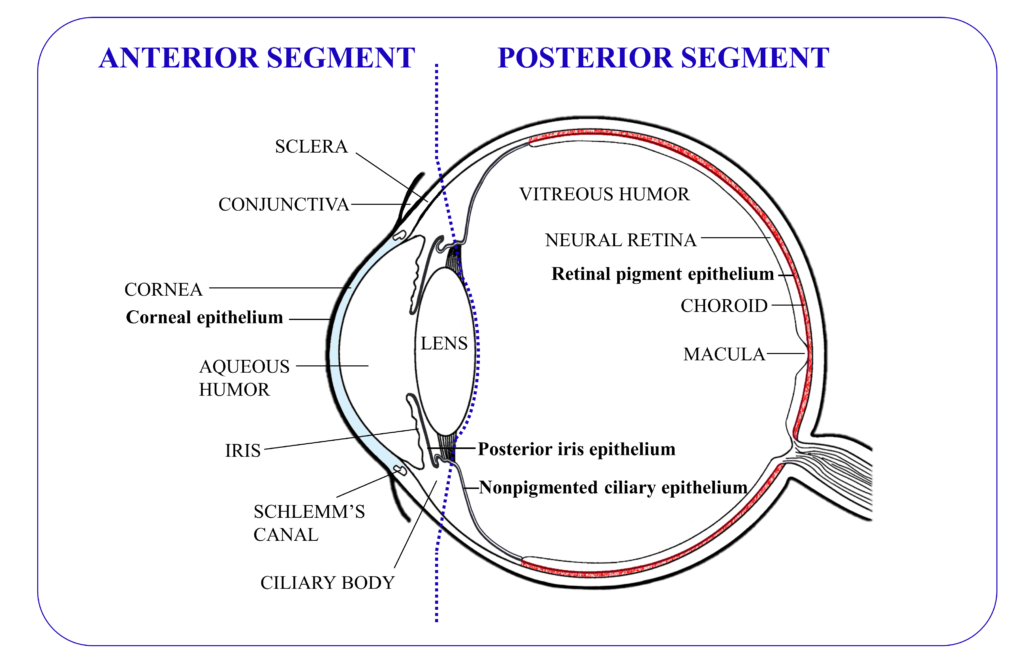
Figure 1: Graphical representation of the anatomy of the eye, with the tissues that contain tight junctions depicted in bold (excepting those in the ocular blood vessels).
Topical administration is the most common and least invasive route of ophthalmic administration, and patients can usually administer the drops themselves. However, eye drops require frequent administration during the day, and are only effective for treating illnesses of the anterior segment of the eye. Additionally, patient compliance is often low, especially for chronic indications such as glaucoma.2
Intravitreal injections are used to treat illnesses of the posterior segment of the eye. This is an invasive route but it is the only one that provides effective drug concentrations to the target tissue. In practice, neither topical nor systemic routes of administrations have proven to be feasible alternatives. Topical administration does not ensure high enough drug concentrations at the back of the eye due to the anterior ocular barriers and flows, whereas systemic administration would require excessively high body-wide drug exposure in order to achieve a meaningful concentration of drug in the retina or choroid, because of the blood-ocular barriers (Figure 2).
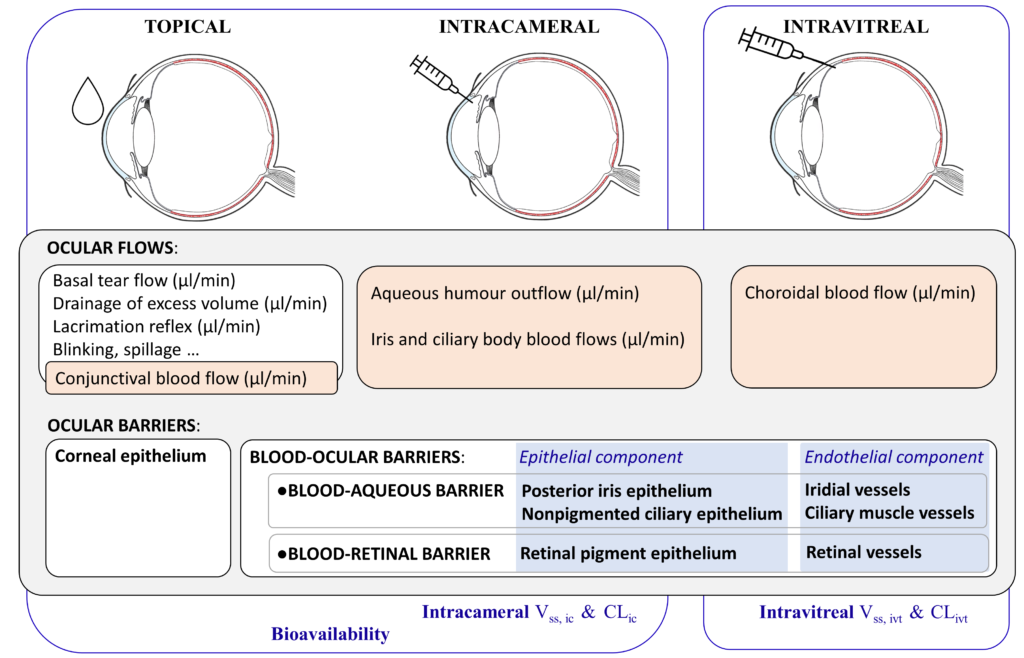
Figure 2: Topical, intracameral and intravitreal routes of administration, the ocular flows and barriers affecting the pharmacokinetics of small drugs, and the corresponding relevant pharmacokinetic parameters: intracameral (ic); intravitreal (ivt); volume of distribution (Vss); clearance (CL).
Typically, small drugs are applied topically, while biologics, such as ranibizumab (fragment antigen binding), aflibercept (soluble receptor) and the recent US FDA-approved faricimab (bispecific antibody), are intravitreally injected. Nevertheless, new technologies aimed at prolonging the residence time of small drugs after intravitreal administration are currently being intensively investigated (other areas of research include topical, intracameral and subconjunctival – between the conjunctiva and sclera – administration). Going forward, this article will focus on small drugs.
“New technologies aimed at prolonging the residence time of small drugs after intravitreal administration are currently being intensively investigated.”
PHARMACOKINETICS
The importance of pharmacokinetics during drug design and development has been established for a long time. The primary cause of attrition of drug candidates in development in the 1990s was inadequate pharmacokinetics,3 and today this still remains a challenge when moving into Phase I and II clinical trials.4
In simple terms, pharmacokinetics can be defined as what the body does to a drug: absorption, distribution, metabolism and excretion. The primary pharmacokinetic (PK) parameters are volume of distribution (Vss, mL) and clearance (CL, mL/h). The Vss is not a physiological volume but a theoretical volume that informs on the ability of the drug to permeate into the surrounding tissues and accumulate in them. The CL is the parameter that describes the efficiency of elimination of the drug from the body. These two parameters, in turn, determine the half-life (t½, h) of the drug according to Equation 1:
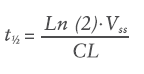
For systemic drugs, the primary PK parameters are obtained after intravenous injection and their value range is well established.5 For ocular drugs there are two possibilities, intravitreal or intracameral administration, both of which have limited data available.6–10 The primary PK parameters can be related and explained by the physiological processes of the eye and are used to design dosage regimens, as well as to define PK models to simulate new data, such as for drugs incorporated in a delivery system.
For intravitreal administration, the blood-ocular barriers and the choroidal blood flow are the key physiological factors controlling the kinetics of these small drugs (Figure 2).
A few years ago, the most extensive and curated collection of primary intravitreal PK parameters of small drugs (40) and biologics (11) to date was published.7 Based on this small drug set, a quantitative structure-property relationships (QSPR) model of intravitreal CL (CLivt) with good statistical values (R2X and R2Y over 0.5) was built, as shown by Equation 2:
LogCLivt (mL/h) = -0.25269 – 0.53747LogHD + 0.05189LogD7.4
HD – Hydrogen bond donor capacity
LogD7.4 – Logarithm of the octanol-water distribution co-efficient at pH 7.4
This model enables the prediction of the CLivt of new drug candidates based on their HD and LogD7.4, if the compound belongs to the same chemical space as the modelled drug set. Indeed, these properties correlate with drug permeability in biomembranes supporting the role of the blood-ocular barriers on CLivt.
Another process that may influence a drug’s CL is metabolism. Metabolism in ocular tissues has only been sparsely studied. Recently, UEF DrugTech has published one of the most comprehensive ocular PK studies including the metabolism of four drugs (acetaminophen, brimonidine, cefuroxime axetil and sunitinib) and two administration routes (intracameral and intravitreal), wherein the concentrations of both parent drug and the main metabolite were analysed in six different ocular tissues.8 It was observed that the impact of ocular metabolism on CLivt and intracameral CL (CLic) seems to be small, except in the case of drugs that are substrates of esterases. Therefore, it was concluded that CLivt and CLic are controlled by the ocular flows and barriers (i.e. by excretion rather than by metabolic processes), CL being permeability limited rather than perfusion limited.
Regarding Vss, ivt, only a very narrow range of values was observed, those being typically one to two times the anatomical volume of the vitreous humour.7 In UEF DrugTech’s recent publication, the lipophilic sunitinib drug showed high partitioning to the surrounding ocular tissues after injection, with the highest Vss, ivt (3.73 mL) ever published, but still only 2.5 times higher than the volume of the vitreous humour.8 The Vss, ic estimated for a narrow set of seven drugs showed a wider range – between two to five times the anatomical volume of the anterior chamber.6,8–10
“With knowledge of the primary PK values of a compound, PK simulations of intravitreal, intracameral, subconjunctival and topical drug delivery systems can be carried out.”
DESIGN OF OCULAR DELIVERY SYSTEMS WITH PK TOOLS
With knowledge of the primary PK values of a compound, PK simulations of intravitreal, intracameral, subconjunctival and topical drug delivery systems can be carried out. Alternatively, QSPR-derived CLivt, and typical Vss, ivt values can be used when new intravitreal molecules are investigated, even before they are synthesised, as only the chemical structure is required to calculate the CLivt.7
The structure of these models is based on compartments or “building blocks” (where the drug is assumed to be well mixed and kinetically homogeneous) that are connected with drug flows (drug amount/unit of time). For example, the input flow into the “vitreal compartment” will be defined by the drug amount loaded in the delivery system and the release rate constant from the “formulation compartment”, while the output flow will be specified by the amount of drug within the vitreal compartment, the volume of distribution and the clearance. Numerous simulations can be carried out to establish the relationships between drug dose, release rate, duration of action and target concentration. Depending on the drug potency (drug concentration required to ensure the desired therapeutic action in the target tissue), it is possible to estimate the required loading dose and make initial decisions, such as the practicability of the ocular formulation (e.g. ensuring the required dose is suitable for a 100 μL intravitreal injection).7,11,12
For instance, the recently reported intravitreal and intracameral PK parameters of brimonidine8 can be integrated into these models to guide the design of an intravitreal insert for a double action treatment for glaucoma:
“The bioavailability of topical ophthalmic drugs is a key PK parameter that enables direct and quantitative comparison of the ocular exposure of topical drugs or formulations. However, this parameter is reported only for a few drugs.”
- Decreasing the intraocular pressure in the anterior segment (via the agonist effect on α2 adrenergic receptors), reducing the production of aqueous humour and increasing its outflow via the uveoscleral pathway.
- Providing neuroprotective action to the posterior segment (optic nerve and retina).
These simulations provide brimonidine concentrations in vitreous and aqueous humour. By considering the dose, release rate constant and therapeutic levels, the reliable duration of a new therapeutic formulation can be estimated. These mathematical tools can advance the development of new ophthalmic formulations, not only for intravitreal administration but also for the intracameral, subconjunctival and topical routes.
Even though UEF DrugTech’s recent study has shown that the role of ocular metabolism in drug CL seems to be of low impact (omitting esterase drug substrates),8 one should observe the low levels of metabolites for all the investigated drugs. For this reason, ocular metabolite toxicity needs to be thoroughly investigated, especially when developing long-acting drug formulations.
BIOAVAILABILITY OF TOPICAL OPHTHALMICS
The bioavailability of topical ophthalmic drugs is a key PK parameter that enables direct and quantitative comparison of the ocular exposure of topical drugs or formulations. However, this parameter is reported only for a few drugs.9,10,13 Bioavailability is calculated as the ratio of drug exposure in aqueous humour after the topical and intracameral administrations of the same drug, exposure (area under the concentration-time curve, AUC0-∞) being dose normalised, as shown in Equation 3:
![]()
Notice that, even though the aqueous humour is not the target tissue, it is the tissue that can be sampled and is therefore the one used for bioavailability calculations (similar to the plasma for systemic drugs). Bioavailability informs on the drug fraction absorbed and can be reliably compared across drugs to evaluate which one is the most penetrating. Nevertheless, more PK studies are required to calculate this parameter for more molecules. In particular, the number of published intracameral PK studies is limited, much more so than intravitreal ones, with only a few CLic, Vss, ic and AUC0-∞, ic values reported in the literature6,9,10 – in particular, the author recommends the literature review presented in Reference 6, excluding the ketorolac and flurbiprofen PK data (the aqueous humour was sampled from the same animal). These PK studies are likely rare due to the difficulty of performing intracameral injections; however, they definitely yield relevant PK parameters for modelling, simulations and quantitating topical absorption data.
“PK models and simulations can guide and inform us when navigating the significant uncertainties encountered during ocular
drug development, guaranteeing a reliable drug- and route-specific framework for the design of drug delivery systems.”
TRANSLATION FROM RABBIT TO HUMAN
Most of the ocular PK information available has been obtained from PK studies in rabbits, which is certainly the case for the data reported in this article. While one can easily argue the validity of the generated PK parameters due to the anatomical differences between the rabbit and the human eye, it is important to notice that models are defined by their purpose and are simplifications rather than exact replications of the system to model. A rabbit eye is not an exact replicate of a human eye but rabbit eyes can be used to predict ocular PK parameters in patients accounting for the key ocular physiological differences for the chosen administration route.
Namely, an extensive investigation on the available intravitreal PK data in humans was undertaken that compared the primary PK parameters between rabbits and humans for small drugs.14 This is the recommended way of analysing the data, as primary PK parameters can be related to the physiology of the eye. Rabbit-to-human CLivt showed a positive correlation.14 The appropriate scaling factor from rabbit-to-human CLivt for hydrophilic small drugs is expected to be approximately 1.4. On the other hand, for the rabbit-to-human Vss, ivt, the scaling factor is closer to three due to the proportional difference between the vitreous anatomical volumes.15,16
More investigation is needed in this area, taking into account the different psychochemical properties of small drugs, other types of drug (i.e. biologics), administration routes and disease states. Nevertheless, these promising preliminary results are encouraging for using rabbits as a relevant ophthalmic PK animal model.
CONCLUSION
PK models and simulations can guide and inform us when navigating the significant uncertainties encountered during ocular drug development, guaranteeing a reliable drug- and route-specific framework for the design of drug delivery systems. The new data generated, PK parameters estimated and the translation factors established offer a solid foundation for future mechanistic models, such as physiologically-based pharmacokinetic (PBPK) models. In PBPK models, the compartments represent real anatomical spaces (tissue volumes) and the drug transfer is based on tissue blood flow, drug partition coefficients and clearances. PBPK models are much more demanding, requiring considerable in vitro and in vivo data for their construction. However, the payoff is greater too, with a greater capability for translating information from preclinical and clinical scenarios and enabling a deeper understanding of the physiological factors and disease effects on drug disposition. In conclusion, all these PK tools significantly benefit the development of ophthalmic drugs and formulations to treat ocular illnesses in both the front and back of the eye.
REFERENCES
- O’Rourke MJ, “The Development and Commercialization of Sustained-Release Ocular Drug Delivery Technologies”. Controlled Release Society Focus Group – Ocular Delivery webinar, Feb 2021.
- Sleath B et al, “The Relationship Between Glaucoma Medication Adherence, Eye Drop Technique, and Visual Field Defect Severity”. Ophthalmology, Dec 2011, Vol 118(12), pp 2398–2402.
- Kola I, Landis J, “Can the Pharmaceutical Industry Reduce Attrition Rates?”. Nat Rev Drug Discov, Aug 2004, Vol 3, pp 711–715.
- Waring MJ et al, “An Analysis of the Attrition of Drug Candidates from Four Major Pharmaceutical Companies”. Nat Rev Drug Discov, Jun 2015, Vol 14, pp 475–486.
- Rowland M, Tozer TN, “Clinical Pharmacokinetics and Pharmacodynamics. Concepts and Applications” 4th Revised Edition. Lippincott Williams and Wilkins, Feb 2010.
- Fayyaz A et al, “Ocular Intracameral Pharmacokinetics for a Cocktail of Timolol, Betaxolol, and Atenolol in Rabbits”. Mol Pharm, Feb 2020, Vol 17(2), pp 588–594.
- del Amo EM et al, “Intravitreal Clearance and Volume of Distribution of Compounds in Rabbits: In Silico Prediction and Pharmacokinetic Simulations for Drug Development”. Eur J Pharm Biopharm, Sep 2015, Vol 95, pp 215–226.
- del Amo EM et al, “Ocular Metabolism and Distribution of Drugs in the Rabbit Eye: Quantitative Assessment After Intracameral and Intravitreal Administrations”. Int J Pharm, Feb 2022, Vol 613, Article 121361.
- Naageshwaran V et al, “Topical Pharmacokinetics of Dexamethasone Suspensions in the Rabbit Eye: Bioavailability Comparison”. Int J Pharm, Mar 2022, Vol 615, Article 121515.
- Naageshwaran V et al, “Comprehensive Ocular and Systemic Pharmacokinetics of Brinzolamide in Rabbits After Intracameral, Topical, and Intravenous Administration”. J Pharm Sci, Jan 2021, Vol 110(1), pp 529–535.
- Subrizi A et al, “Design Principles of Ocular Drug Delivery Systems: Importance of Drug Payload, Release Rate, and Material Properties”. Drug Discov Today, Aug 2019, Vol 24(8), pp 1446–1457.
- del Amo EM et al, “Pharmacokinetic Aspects of Retinal Drug Delivery”. Prog Retin Eye Res, Mar 2017, Vol 57, pp 134–185.
- Fayyaz A et al, “Topical Ocular Pharmacokinetics and Bioavailability for a Cocktail of Atenolol, Timolol and Betaxolol in Rabbits”. Eur J Pharm Sci, Dec 2020, Vol 155, Article 105553.
- del Amo EM, Urtti A, “Rabbit as an Animal Model for Intravitreal Pharmacokinetics: Clinical Predictability and Quality of the Published Data”. Exp Eye Res, Aug 2015, Vol 137, pp 111–124.
- Green H, Sawyer JL, Leopold IH, “Elaboration of Bicarbonate Ion in Intraocular Fluids II. Vitreous Humor, Normal Values”. AMA Arch Ophthalmol, 1957, Vol 57(1), pp 85–89.
- Azhdam AM, Goldberg RA, Ugradar S, “In Vivo Measurement of the Human Vitreous Chamber Volume Using Computed Tomography Imaging of 100 Eyes”. Transl Vis Sci Technol, Jan 2020, Vol 9(1), Article 2.