


Citation: Parry M, “In Vitro Bioequivalence – Where are we Now?”. ONdrugDelivery Magazine, Issue 102 (Nov 2019), pp 38-40.
With a focus on bioequivalence testing in generic inhalables development, Mark Parry highlights shortcomings of aerodynamic particle size distribution and delivered dose testing, and introduces newer testing techniques.
In vitro bioequivalence for the development of generic inhaled pharmaceutical products continues to present industry with significant and evolving challenges. Developing and performing suitable analytical strategies to satisfy evolving regulatory requirements – whilst establishing a robust development process which ensures that the developed generic product is successful – continues to highlight the limits of the traditional approaches to in vitro characterisation.
Regulators are advancing the expectations for the content of in vitro testing programmes through continued development of general and product-specific guidance. These expectations include newer techniques such as spray pattern plume geometry testing for metered dose inhalers – and a clearer focus on identifying and controlling critical quality attributes (CQAs) as an integral part of the overall development.
The traditional inhaled product performance tests of aerodynamic particle size distribution (APSD) and delivered dose have proven themselves to be vital tools in the development and testing of inhaled products and form the backbone of any inhaled product specification. We must, however, understand the shortcomings of these tests. While they can tell us information about how much drug is released and its size, they do not provide information on other factors such as the nature of the deposition (e.g. individual particles, agglomerates or co-deposited with a carrier or a second API particle) or the morphology of the particles themselves. Both are factors that will influence actual bioavailability and delivery of the drug locally and systemically.
Many companies have been in the position where in vitro work has delivered a strong and comprehensive data package showing good equivalence between the reference and generic products, only to see pharmacokinetics or pharmacodynamics in vivo data show unacceptable differences between the products. Clearly, we need more physiologically relevant data for our in vitro analysis and, while standard APSD and delivered dose tests themselves are not accurate physiological models, they do provide a platform we can build on.
NEW TECHNIQUES
Several advanced techniques are now available which can provide useful input to improve the ruggedness of the development programme to de-risk clinical studies or to bridge in vivo-in vitro correlation (IV-IVC), thus strengthening in vitro data submissions with a view to potentially avoiding clinical work.
Flow Profiles
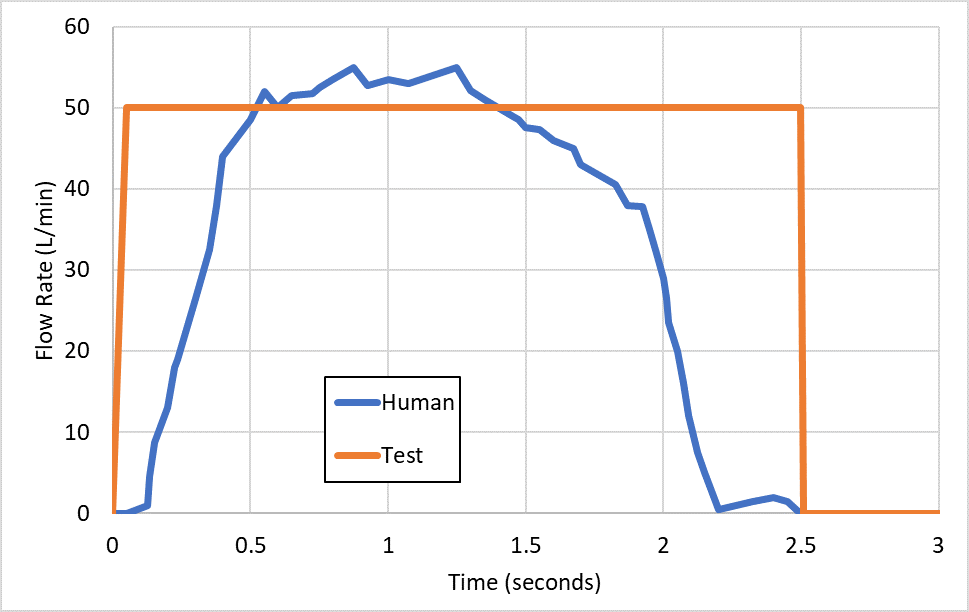
Figure 1: Differences between human and test flow profiles.
Standard APSD and delivered dose testing make use of simplified flow profiles for testing, effectively a square wave where the flow is on or off, at a physiologically relevant flow rate. For dry powder inhalers (DPIs), the air flow is critical as it provides the energy for delivering the drug from the device itself. However, a square wave is not an accurate reflection of a normal human inhalation flow profile.
Current DPI regulatory guidance does acknowledge the need to look at a range of flow rates for the reference and generic product to explore the robustness of the product to the varying maximum flow rates likely to be seen in any typical population. Nevertheless, normal conditions do not provide an effective simulation of the actual likely behaviour observed in both healthy and inhalation-compromised users, where a range of flow profiles will be seen.
Particular attention to the impact of the patient’s condition on the typical inhalation manoeuvre observed can provide for a more biorelevant test that can be incorporated into the APSD and delivered dose testing – and provide data likely to be more clinically relevant. An example of a normal human profile versus the test profile is given in Figure 1.
Lung Dissolution
While impaction testing provides good information on the aerodynamic size of the inhaled product, it does not provide information on the likely behaviour of the drug observed at the various particle sizes when deposited in the lung. The dissolution and absorption of the API can be affected by factors beyond just its size, with the particle morphology, salt and crystal form all likely to have an impact.
In the same way that traditional dissolution tries to mimic the dissolution of tablets and capsules in the stomach, lung dissolution testing aims to do the same thing for inhaled products. There are, however, several factors to address.
First, we need to identify a method of preparing the sample. Modified impactor stages and similar techniques present a way of isolating the relevant size particles as we only want to test particles that can deliver to the lung. We then need to identify a suitable medium, with simulated lung fluids requiring a complex mix of salts and other components to mimic expected behaviour, and then a suitable equipment approach is needed. This can involve the use of traditional dissolution vessels with suitable holders for the collected particles. However, other options are being explored as well.
Work continues across the industry to develop and optimise lung dissolution models to allow for the dissolution behaviour of the delivered API to be assessed. While there may not yet be a harmonised method available, these techniques provide a better way to model a drug’s bioavailability and are proving to be an increasingly important part of generic product development.
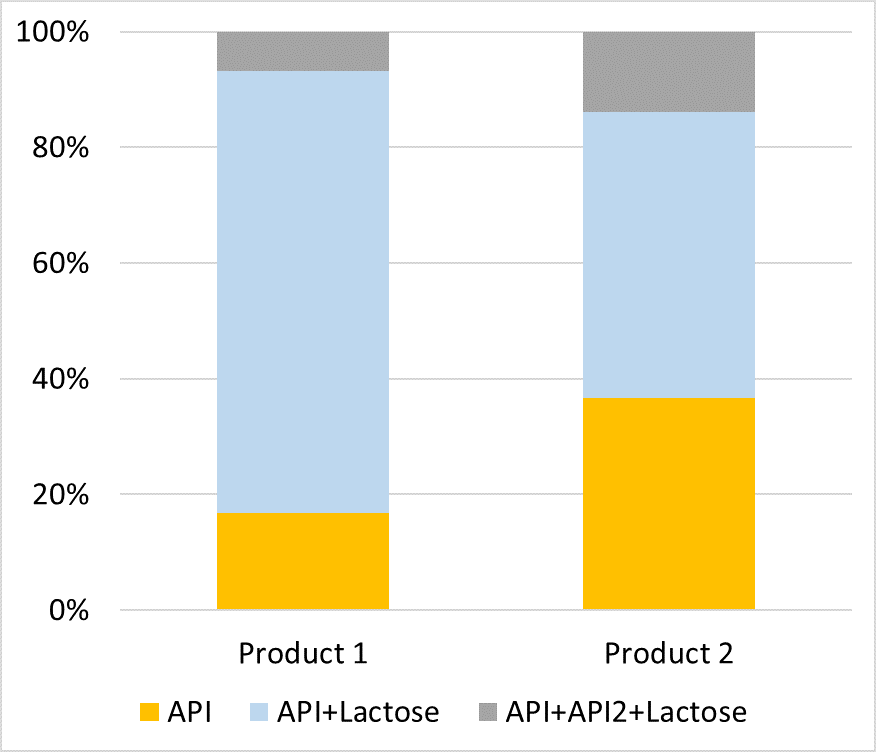
Figure 2: The different deposition behaviour of API particles in two products at one size fraction.
Morphologically Directed Ramen Spectroscopy
Recent work looking at morphologically directed ramen spectroscopy (MDRS) provides evidence for a useful orthogonal technique to complement impaction. This technique combines optical microscopy, computerised particle measurement and per-particle ramen spectroscopy to deliver information on the size, shape and identity of individual particles.
Using modified NGI stages we can examine individual size fractions and information regarding particle morphology to be generated and compared. Further development of the technique also allows for assessment of the product’s deposition behaviour, allowing information regarding the deposition and distribution of API only, API/carrier, API1/API2 and API1/API2/carrier particles to be directly measured – which was not previously practical. See Figure 2 for an example of the differences in API particle deposition for two products at the same size fraction.
Having this data provides vital information on both the morphology and deposition behaviour of the product and allows for further differentiation in performance of products – even when traditional drug mass data may suggest they are equivalent.
INTERTEK’S SOLUTIONS
Given the challenges in delivering successful clinical bioequivalence studies – even with acceptable in vitro data – recent expansion at Intertek’s Centre of Excellence for Inhaled and Nasal Drug Development in Melbourn, UK, has focused on new, powerful in vitro analytical strategies. The expansion of our laboratory footprint also enables us to help our clients’ key decision-making activities throughout the product development lifecycle.
We understand the need to invest time to establish rugged methodology with a focus on identifying and controlling CQAs as an integral part of product development. Intertek’s experienced scientists deliver in vitro analytical programmes to support all stages of development for both innovator and generic products as well as maintaining involvement in development of new and improved techniques and technologies. Looking ahead, these new analytical strategies should allow for the development of stronger in vivo data packages supporting both greater clinical success and the possibility of successful in vitro only generic approvals.


