

To Issue 150
Citation: Riter J, Morgan V, “Successful Industrialisation Requires Solid Foundations.” ONdrugDelivery, Issue 150 (Jul 2023), pp 19–22.
Jennifer Riter and Victoria Morgan discuss the importance of choosing the right containment system at the start for successful industrialisation of an injectable drug product.
“Formulation, materials, machinery, technology and human factors must all be synchronised in harmony.”
In recent years, pharmaceutical manufacturers and regulatory authorities have increased their scrutiny of the supply chain’s ability to deliver even greater levels of quality – all in the expectation of delivering improved patient safety and compliance. This expectation is against a backdrop of more complex and costly drugs being developed, as well as an ongoing drive for greater efficiency in manufacturing.
To address these complex issues, containment system and drug delivery system manufacturers have developed components that not only ensure quality, safety and efficacy throughout a drug product’s lifecycle, they also mitigate the risks and maximise the efficiencies of fill-finish processing.
In this article, we will discuss how choosing the appropriate quality of containment systems from the outset is critical to the successful industrialisation of an injectable drug product. We will review some of the challenges associated with injectables – most notably extractables, leachables and particulates – which are some of the primary reasons for a product recall.
We will then showcase how West Pharmaceutical Services has addressed these challenges with an integrated offer of NovaPure® components, coupled with analytical testing services. Together, this portfolio delivers pharmaceutical manufacturers the reassurance they need that extractables and leachables, particle analysis, container closure integrity, product performance and processing considerations have been addressed appropriately and that they have the data they need for a successful approval.
We will then review the implications of the European Medical Device Regulation (MDR), specifically regulation 2023/607, for pharmaceutical manufacturers, particularly in terms of the data needed. Finally, we will present a case study on how West’s integrated technical document package can assist in reducing the time, cost and risks associated with MDR submissions.
ENORMOUS COMPLEXITY
It might be argued that the simplistic terminology within drug development is failing us. Talk of pathways, roadmaps and journeys accurately conveys the process, but these succinct terms are truly insufficient to express the enormous complexity involved in guiding a molecule from research and development through to regulatory approval and manufacture at scale.
The ultimate goal of industrialising a product requires pharmaceutical manufacturers to orchestrate a range of variables into a delicately co-ordinated and closely managed process to realise the associated economic benefits. Formulation, materials, machinery, technology and human factors must all be synchronised in harmony. At the same time, however, each one of these elements has the potential to undermine the entire process and, therefore, to compromise the output.
For a drug product, any such compromise can present a danger to patient safety and introduce the possibility of a recall, which carries both financial and reputational consequences. When the process is industrialised at scale, such risks are amplified. The same multiplier effect that allows productivity gains to be unlocked can mean the damage is exponentially more significant.
“The plunger plays a critical role because it is in direct contact with the drug formulation.”
MITIGATING THE RISK
Managing the quality of every element in an industrialised process is therefore crucial to mitigate the risk of any problems occurring, either at the point of development, during production or later in the product’s lifecycle. Integrated combination products, such as prefilled syringes (PFSs), already address many risk-mitigation factors by delivering an accurately measured ready-to-administer dose of a drug product in single-use form. This reduces the risk of human error associated with administration, while also avoiding the possibility of coring of the elastomer stopper when it is punctured by a needle, which can inherently cause the patient to not get the appropriate dose of their drug product.
Maintaining drug integrity within a PFS places particular emphasis on the primary packaging components that will remain in contact with the drug product following fill-finish and on into transportation and storage. Among these, the plunger plays a critical role because it is in direct contact with the drug formulation, meaning there is an imperative for this part of the containment system to demonstrate qualities that will avoid, or significantly limit, the potential for formulation integrity to be compromised. Any impairment in terms of purity, sterility or stability can, in turn, impair the product’s efficacy and safety.
West’s NovaPure plungers have been designed and manufactured using quality by design (QbD) principles to address these risks and meet the exacting requirements for performance and patient safety. In the case of NovaPure plungers, these proven benefits are realised through a combination of the enhanced design, the elastomeric formulation and the FluroTec™ barrier film.
The specification of a component such as a NovaPure plunger ensures a key building block is in place for industrialised production, but components cannot be considered in isolation when establishing a quality-based, large-scale manufacturing process. This also relies heavily on testing and evaluation to verify the sustained compatibility between the drug product and containment system from both a physical and chemical perspective. Among the main objectives is providing evidence that extractables, leachables and particulates are kept to an absolute minimum while container closure integrity (CCI) is securely maintained (Figure 1).
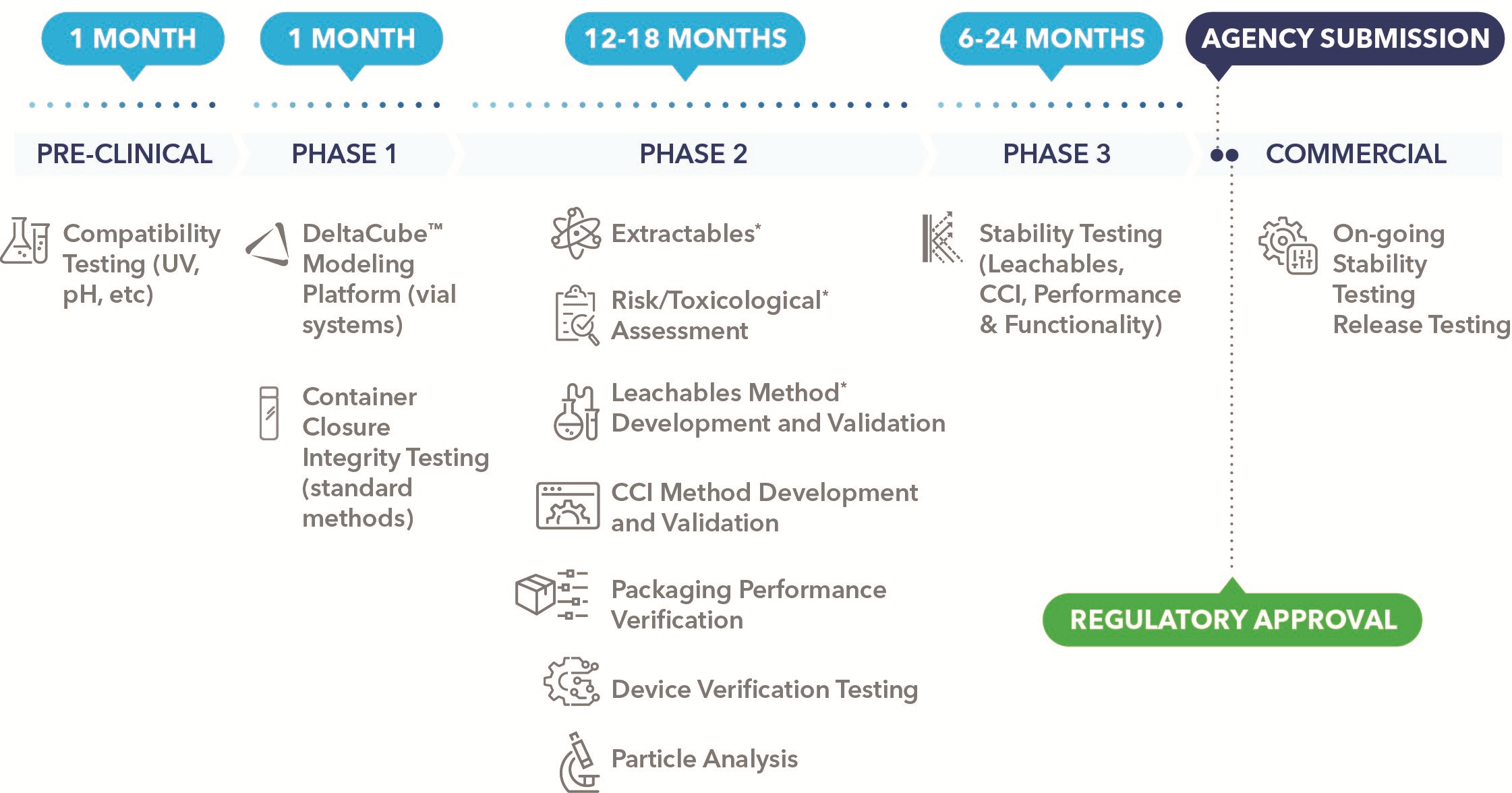
Figure 1: Planning drug product packaging testing with drug product development can help reduce delays to your regulatory submission. *Extractables, risk/toxicological assessment and leachables are performed sequentially and therefore require the greatest amount of time of 18 months.
ANALYTICAL TESTING
At West, analytical testing services are delivered as part of a wider quality-focused approach, complementing components such as NovaPure plungers to ensure compatibility between the packaging system and drug product is evaluated at an early stage and will minimise any risk or concerns when production is scaled. Working closely with pharmaceutical manufacturers, West provides an initial assessment of the risk of extractables and leachables – and develops a comprehensive series of analytical tests that will elicit data on any continued risk presented by these elements over the shelf life of a drug product.
Parallel analysis will be carried out to test the CCI of the PFS system. This is achieved using a variety of deterministic methods, including high-voltage leak detection, to provide pharmaceutical partners with a comprehensive, data-backed understanding of the materials and components that comprise the system, as well as the mechanics of how they function together to protect against issues such as leakage and ingress over time. Separate analytical testing will also be carried out to assess the risk of contamination from particulates, evaluating the potential for packaging components, manufacturing processes or extrinsic environmental elements to result in the presence of visible or sub-visible particles within the contained dose.
The provision of such extensive analytical testing methods, dovetailed with the supply of quality products, affords pharmaceutical manufacturers a holistic view of the entire drug containment system. It provides a platform to evidence data and compliance with regulatory standards, such as MDR, at all levels – from components through to the finished product. Crucially, there is also consideration from the very beginning of how those standards can be met in the context of scalability and the continued need to apply rigorous quality control during volume production.
“Because of its rigorous nature, the process of assessing conformity is not necessarily a straightforward one.”
COMPLIANCE
The process of demonstrating compliance has, of course, become more involved for pharmaceutical companies in recent years as regulators tighten controls around product quality and patient safety. In the EU, for example, the EU MDR (2017/745/EU), first published on April 5, 2017, introduced several changes designed to safeguard device quality. This was implemented in the wake of the PIP breast implant scandal, where the fraudulent manufacture of implants using unapproved silicone gel left many patients at increased risk of rupture.1 Compared with the EU directive it replaced (93/42/EEC), the MDR places far greater emphasis on patient safety and sets out a far more comprehensive set of enforced requirements for pharmaceutical manufacturers to follow.
Implementation of the regulation was already subject to a phased transition before being disrupted by the pandemic in 2020 and finally coming into force on May 26, 2021. Subsequent challenges, however, including the increased demands placed on marketing authorisation holders and the need for capacity-limited notified bodies to fulfil a more involved role in device approvals, led the European Commission to propose extensions to the transition periods stipulated in Article 120 of the regulation. For Class IIB non-implantable devices, Class IIA and Class I devices, which encompasses PFSs, this now means the deadline for transition has been extended to December 2028.
This “relief” has been balanced with stringent conditional requirements to actively implement compliance measures. This includes the need for a device manufacturer to establish a quality management system (QMS) by May 26, 2024 and for that QMS to be audited and approved as compliant by the relevant notified body no later than September 2024. Conformity assessments carried out by notified bodies incorporate a detailed review of technical documentation compiled to evidence a device’s general safety and performance requirements (GSPRs) as well as interrogation of the clinical evaluation report, which contains testing data on the clinical use of a device. As part of a wider information-gathering process, these documents are reviewed in light of Annex I of the revised MDR to inform the risk-based assessment conducted by the notified body.
ASSESSING CONFORMITY
Because of its rigorous nature, the process of assessing conformity is not necessarily a straightforward one, and this can result in delays to the marketing authorisation holder receiving a notified body opinion. Problems can relate to incomplete submissions, where there is insufficient information provided to demonstrate compliance with MDR, or simply the fact that the supplied technical documentation is poorly structured, which complicates the notified body’s already arduous task of appraising all the supplied materials.
At West, a commitment to QbD principles means it can support pharmaceutical partners at each stage along this updated and extended approval pathway. The company’s knowledge of the process and the nature of the information and data required puts it in an ideal position to compile and co-ordinate all necessary technical information relating to its components for efficient filing in a timely manner. In streamlining this process, West’s overarching objective is to empower pharmaceutical manufacturers in their engagement with a notified body and, ultimately, the EMA, ensuring all documentation and certification is available to demonstrate compliance with the updated regulatory framework.
TECHNICAL DOCUMENT PACKAGE
This service offering has been encapsulated as the Technical Document Package (TDP). Borne out of first-hand experience, it comes after West conducted a successful pilot programme to support a pharmaceutical customer with an MDR filing for a new drug product delivered via a PFS system incorporating a NovaPure plunger. The aim of the programme was to address the recognised challenges associated with the filing process, including the need for cross-organisational stakeholder management and the co-ordination of consistent responses.
Working closely together with the customer, West adopted a strategic approach to gathering, assimilating and presenting the required data at a level of detail and in a format that would satisfy scrutiny by the notified body. By focusing efforts on the “right-first-time” delivery of high-quality and highly relevant information connected to the specific GSPRs, West was able to minimise requests for supplemental information, avoid complex multi-document iterations and truncate the entire certification process.
In turn, this positive outcome has given West a robust platform from which to develop a transferrable template for supporting a successful filing in a time-efficient manner in the form of the TDP. This offering currently supports applications for 1–3 mL NovaPure plungers by incorporating more than 30 documents, including component technical summaries, quality statements and compliance bulletins, into a single, integrated information package. Future developments will see the offering extended further into West’s range of leading primary packaging components.
The changes to MDR, while specific to the EU, offer an indication of the continual drive among regulatory bodies to address novel technologies and ensure patient safety. For pharmaceutical manufacturers navigating the many challenges involved in bringing an integral drug product to market at industrial scale, the TDP provides a reassuringly efficient platform to demonstrate compliance. Furthermore, it provides an effective reminder that, whether in the provision of individual components or supporting analytical services or entire drug delivery systems, a strategy rooted in quality can allow risks to be controlled and production to flourish.
At West, this quality philosophy has been deep rooted for decades, a point perhaps best illustrated by the fact that all the top 10 best-selling global injectable drugs in 2022 used West packaging.
NovaPure, FluroTec and DeltaCube are trademarks or registered trademarks of West Pharmaceutical Services, Inc. FluroTec technology is licensed from Daikyo Seiko, Ltd.
REFERENCES
- “PIP breast implants”. Web Page, UK NHS. (www.nhs.uk/conditions/pip-implants, Accessed, Jun 1, 2023).
