
To Issue 176
Citation: Raverdy-Wilson S, “Device Design to Address Usability and Mitigate Use-Related Risks – a Medical Affairs and Human Factors Perspective”. ONdrugDelivery, Issue 176 (Sep 2025), pp 28–33.
Dr Sylvine Raverdy-Wilson deep dives into the device constituent of drug-device combination product design and shares perspectives through the Use-Related Risk Analysis methodology with a focus on the BD Libertas™ Wearable Injector.
As the global population ages and chronic diseases become more prevalent, healthcare systems are facing mounting pressure to reduce the burden of care delivery. The demand for patient-centric care models that extend beyond traditional clinical settings is also accelerating. To meet these shifting needs, medical device and pharmaceutical companies are developing drug delivery solutions and drug-device combination products designed for the safe and effective delivery of biologics to enable a transition from intravenous (IV) infusion to subcutaneous (SC) injection or direct development of SC solutions. For the patient, these products may be designed for treatment flexibility and accessibility, potentially enabling care in both clinical and alternative care settings. In all cases, the drug and its delivery system are required to meet stringent safety and efficacy standards, supporting effective outcomes for patients and providers alike.
Meeting these evolving needs requires pharmaceutical companies and their partners to explore new approaches on how therapies are administered (including the possibility of self-administration) while carefully considering the key factors that influence effective delivery of the drug, such as concentration, viscosity and dose volume. While self-administration of dose volumes up to 2.25 mL is commonly facilitated by prefilled, handheld autoinjectors, these injectors may be suboptimal for the SC delivery of larger-volume biologics with longer injection durations, especially in non-traditional care settings.1 In such cases, larger-volume wearable injectors are an opportunity for administration of single bolus doses greater than 2 mL.
“WEARABLE INJECTORS ARE MEDICAL DEVICES OR DEVICE CONSTITUENTS DESIGNED TO ADMINISTER LARGE-VOLUME SC DRUG FORMULATIONS OVER DURATIONS
Wearable injectors are medical devices or device constituents designed to administer large-volume SC drug formulations over durations ranging from minutes to hours. These device constituents, combined with drug formulation, enable the transition from IV to SC delivery. The diversity of device designs available or in development2 is extensive and provides both healthcare providers and patients with a choice that can meet their individual needs.
The development of a drug-device combination product, including its formulation, is a complex, lengthy and costly process. Initially, the drug molecule undergoes rigorous laboratory testing as part of its preclinical drug discovery and development process. This is followed by multiple clinical studies to demonstrate safety and efficacy and to determine the appropriate dosing regimen, volume and frequency required to achieve therapeutic benefit.3 Throughout this process, it is important to anticipate the possibility of using a delivery device and then ensure a seamless integration within a drug-device combination product. It is good practice to select a device constituent whose potential impact on patient safety has been minimised to the greatest extent possible. This approach aligns with regulatory expectations and the risk management standards applicable to combination products.
RISK MANAGEMENT FOR DRUG AND DEVICE CONSTITUENTS
Ensuring the safe and effective use of device constituents within the drug-device combination product requires comprehensive risk management strategies throughout the combination product’s life cycle. Figure 1 is a graphical representation of the risk management cycle for the device constituent (per ISO 14971:2019) and how it integrates with the drug risk management cycle (following ICH Q9 (R1)) to result in the overall drug-device combination product risk evaluation. As such, it is important to proactively control and mitigate risks prior to integration of the device constituent to avoid a negative impact on the overall drug-device combination production risk evaluation.
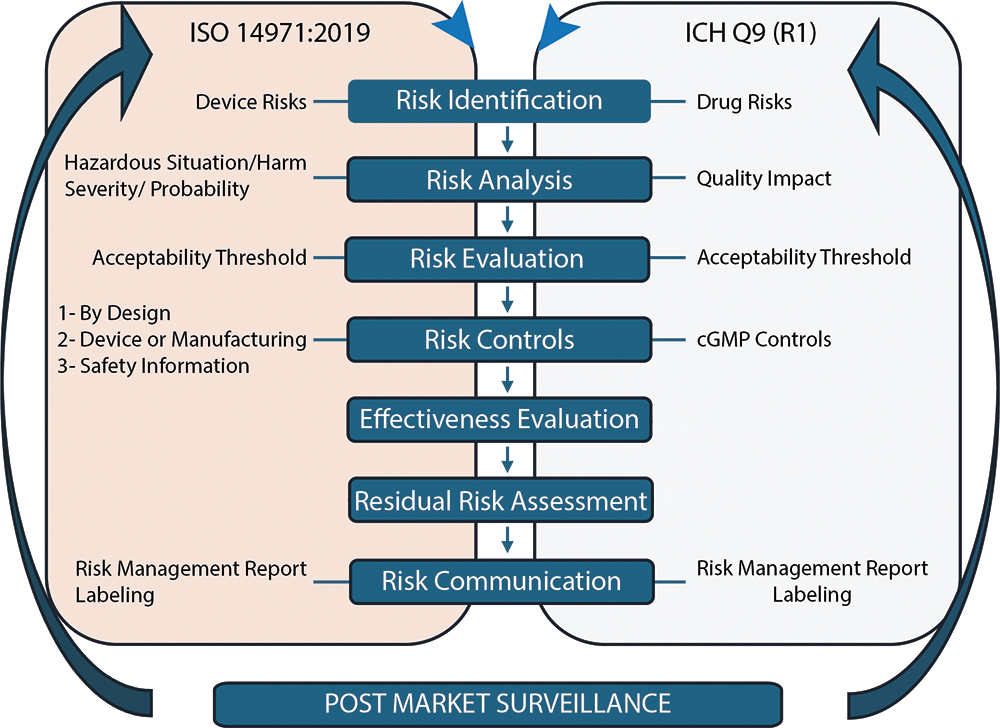
Figure 1: Risk management cycle for medical devices and drugs based on ISO 14971:2019 and ICH Q9 (R1).
ISO 14971:2019 lists the three risk-control mechanisms available to manufacturers to reduce identified risks, with the following priority order:
- Inherently by design
- Through the medical device itself or its manufacturing (barrier or alarm) and, finally
- Through important safety information provided to the patient in the Instructions For Use (IFU).
According to the ISO 14971:2019 standard for medical device risk management, high-level residual risks must be mitigated “as far as possible without adversely affecting the benefit-risk ratio” of the medical device. As a best practice, manufacturers of device constituents should evaluate how their devices will be used across the intended patient populations and indications to ensure seamless integration into the final combination product. Throughout the injector’s use by the patient, caregiver or healthcare provider, there are multiple opportunities for hazardous situations to occur, impacting both the patient and the drug they receive. Evaluating the occurrence of these hazardous situations and the severity of the harm allows for the assessment of residual risk.
While user compliance with instructions and proper technique is essential across all injector designs for combination products, some technologies rely more heavily on these “user-directed controls”. Manual steps – for example, drug transfer using a separate syringe, surface disinfection with antiseptic wipes or other manual assembly steps such as drug cartridge insertion – introduce opportunities for use error. The effectiveness of “user-directed risk controls” depends on multiple factors, including user compliance with labelling, clarity of the IFU, and consistency of technique at first treatment and subsequent treatments when treating chronic diseases. These challenges are further compounded in real-world settings, where distractions and interruptions may compromise user performance, particularly in chronic treatment scenarios.
USE-RELATED RISK ANALYSIS
The Use-Related Risk Analysis (URRA) draft guidance was formally issued by the US FDA in 2024,4 but the tool was previously described in the 2016 FDA draft guidance “Human Factors Studies and Related Clinical Study Considerations in Combination Product Design and Development”.5 It has also been mentioned in several guidances issued by the Center for Devices and Radiological Health, the Center for Drug Evaluation Research and the Center for Biologics Evaluation and Research.6,7
The URRA is a document that systematically analyses each use step of a medical device or drug-device combination product, along with the associated clinical risks. It is a suggested and recommended component of FDA submission packages, including INDs, NDAs and BLAs. It links potential use errors at each step to their clinical risk. Conducting this analysis early in development supports informed decisions on risk-mitigation strategies. The FDA recommends that the URRA includes detailed evaluation methods of the effectiveness of the risk controls during human factors validation testing. Figure 2 summarises the steps of a complete URRA.
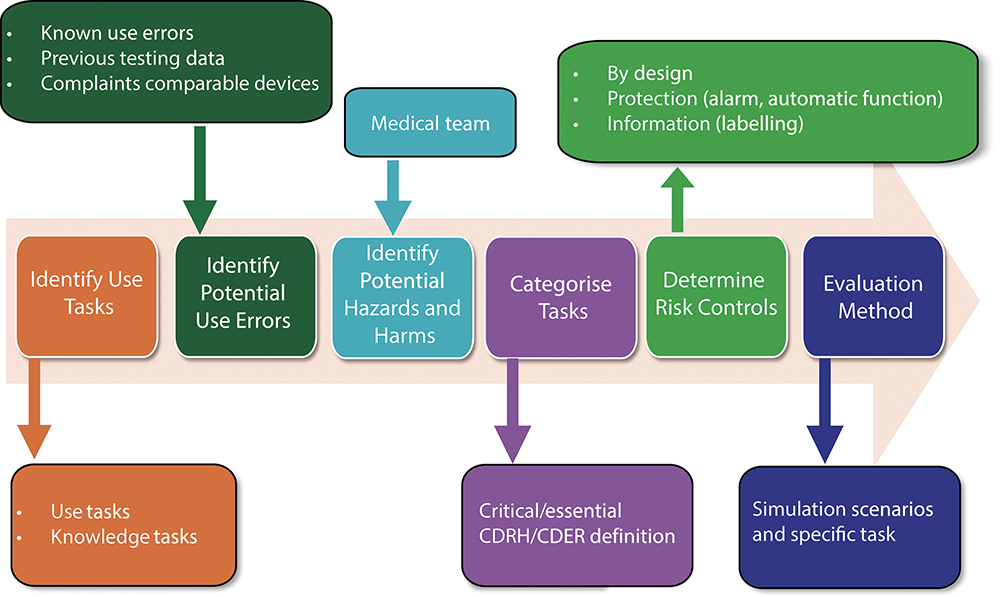
Figure 2: Graphical representation of a URRA per FDA guidance (draft, 2024)4.
APPLICATION OF URRA METHODOLOGY TO WEARABLE INJECTOR USE
There are generally three technology options (commercially available with drug or in development) for administering drug with a wearable injector device constituent. These technology options, represented in Prašnikar et al,2 involve varying preparation steps for patients or caregivers immediately prior to treatment, depending on the technology. The technology options are:
- Transfer Drug from Vial or Syringe to Wearable Injector: Requires patient or caregiver to fill a syringe with drug from a vial and transfer the drug to the wearable injector prior to treatment.
- Loading Drug-filled Container into Wearable Injector: Requires patient or caregiver to clean and load a prefilled drug cartridge into the wearable injector prior to treatment.
- Prefilled, Pre-assembled Wearable Injector: Wearable injector does not require filling or assembly by patient or caregiver prior to treatment.
A simplified analysis was conducted by the author, based on the FDA URRA draft guidance (2024). This analysis focused on the applicability of the steps for use of the third technology. The analysis evaluated the potential effect of mitigating use-related risks through design based upon the range of potential usage steps required when using a wearable injector technology. The BD Libertas™ Wearable Injector is a prefilled, pre-assembled device component designed to deliver single-dose SC injections of large-volume (2–5 or 5–10 mL) and high-viscosity (up to 50 cP) fixed-dose biologics, and it is representative of the third technology.
This paper evaluated user interaction, handling complexity and potential clinical risks associated with wearable injector technologies, with a focus on the BD Libertas™ Wearable Injector. The steps followed in this illustrative analysis were:
- Use Step Identification: IFUs from three wearable injector technologies were reviewed to inform the range of potential usage steps required when using a wearable injector technology. Use steps were grouped by functional category (e.g. warming the package, preparing the injection site). While the order of steps may vary, the core actions are consistent across devices.
- Use Error Identification: One representative potential use error was selected per use step to simplify the comparison.
- Clinical Risk Assessment: Each potential use error was linked to the most direct and plausible clinical harm that could affect both general and immune-compromised patient populations.
Severity ratings, criticality assessments and evaluation of risk control effectiveness were excluded from the analysis. Design and manufacturing-related risks were also excluded.
The analysis was not exhaustive in nature, but it was intended to represent the URRA principles and the value of URRA in the development of a drug-device combination product. The analysis only covered the use-related risk. Results of the analysis are not indicative of clinical performance or outcomes.
Table 1 summarises the information and key findings related to user handling, preparation complexity and potential risk associated with a delivery system.
| Use Steps8,9 | Potential Use Error | Clinical Risk | |
| Warm the injector package | User does not wait the appropriate time | Painful injection | |
| Gather all supplies and wash hands | User fails to wash hands properly | Contamination leading to infection | |
| Inspect the drug/device | User does not detect degraded drugs | Compromised treatment | |
| Injector preparation | Clean drug container (vial or cartridge) | Inadequate cleaning of stopper | Use-related contamination leading to infection Not applicable to pre-assembled injector |
| Transfer drug into intermediate container (drug in vial) |
Incorrect filling technique, air bubbles |
Incorrect dosage, potential overdose or underdose Not applicable to prefilled injector |
|
| Assemble cartridge and injector/Fill injector with drug | Spillage/breakage or contamination during transfer |
Use-related contamination leading to infection Not applicable to prefilled injector |
|
| Unlock the device | User does not know how to unlock the device |
Delay in therapy | |
| Select the injection area | Incorrect site selection | Ineffective delivery | |
| Clean the injection area | Inadequate cleaning of injection area | Infection at injection site |
|
| Apply injector | User error in application | Pain, improper delivery, delay in treatment | |
| Injection | Activate the injector | User does not understand how to start the injection | Delay in therapy |
| Monitor injection progress | User does not understand how to track progress | Confusion or annoyance | |
| Confirm end of injection | User removed the injector too early | Lower dosage | |
| Remove the injector from the skin | User pulls injector too hard from skin | Pain, annoyance | |
| Dispose of the injector | User throws away the injector in regular trash |
Third party exposure to used injector | |
Table 1: Potential use error and clinical harm associated with combination product steps and how they apply for the BD Libertas™ Wearable Injector.
“THE BD LIBERTAS™ WEARABLE INJECTOR FEATURES A
PREFILLED AND PRE-ASSEMBLED DESIGN THAT HELPS TO REDUCE HANDLING STEPS, THEREBY SIMPLIFYING THE PROCESS AND LOWERING THE POTENTIAL FOR USER ERROR, CONTAMINATION AND DOSAGE INACCURACIES.”
RESULTS FOR GENERAL PATIENT POPULATION
The analysis for applicability of the use-related risks associated with combination product use steps for general patient population highlights the varying levels of complexity, contamination risk and dosage accuracy depending on the technology used. This complexity may increase the likelihood of contamination and discrepancies in dosing. The BD Libertas™ Wearable Injector features a prefilled and pre-assembled design that helps to reduce handling steps, thereby simplifying the process and lowering the potential for user error, contamination and dosage inaccuracies.
Overall, by reducing the number of use steps and mitigating by design the potential risks of contamination and dosage issues, the BD Libertas™ Wearable Injector aims to offer a user-friendly design by design, providing a safe and consistent experience for general patient population.
RESULTS FOR IMMUNOCOMPROMISED PATIENT POPULATION
The immune system has two functions – to recognise foreign elements and to defend the body against infection. Several causes can lead to a compromised immune system, such as genetic mutation or a disease, resulting in a higher risk of infection for immunocompromised patients. When comparing the summary of risks for immunocompromised patients with that for general patients, it becomes evident that the heightened vulnerability of immunocompromised patients amplifies the potential consequences of user errors and contamination. The BD Libertas™ Wearable Injector, by mitigating the risk by design, seems to be a suitable drug-device combination product from the use-related risk standpoint for both patient populations.
CONCLUSION
Risk management standards, such as ISO 14971:2019, clearly state that mitigation of risk through design is the highest priority of the three available options (1 – design, 2 – alarm, 3 – IFU). The URRA required by the FDA for medical device and drug-device combination products is a tool that can be used during design development of a medical device or device constituent to identify risk-control measures that will have the most impact on the use-related risks identified.
This paper exercise highlights that reducing the number of use steps can lead to an overall reduction in use-related risks. This aligns with FDA’s 2016 guidance on Applying Human Factors and Usability Engineering to Medical Devices,10 which states: “Design modifications to the device and its user interface are generally the most effective means for eliminating or reducing use-related hazards. If design modifications are not possible or practical, it might be possible to implement protective measures through labeling. These strategies are not the most preferred, though, because they rely on the user to remember or refer back to the information, labeling might be unavailable during use, and knowledge gained through training can decay over time.”
The analysis highlights the importance of drug delivery systems that prioritise ease of use and risk reduction, especially for vulnerable populations, such as immunocompromised patients. One of the inherent benefits of the BD Libertas™ Wearable Injector is its prefilled and pre-assembled design, which simplifies dose preparation and eliminates the potential for breakage, use-led contamination, and dosing errors associated with end-user filling and/or assembly of the drug delivery system at the time of use.
The BD Libertas™ Wearable Injector is designed in accordance with fundamental principles of risk management outlined in ISO 14971:2019 by featuring a prefilled,pre-assembled format. Its Peel, Stick & Click™ mechanism simplifies the injection process, minimising user error and infection risk by shifting preparation responsibilities to the pharmaceutical manufacturer. This not only enhances consistency and reliability in treatment but also reduces the burden on patients. By focusing on user-centric innovation, BD continues to advance drug delivery solutions that support both patient safety and pharmaceutical success.
BD Libertas™ Wearable Injector is a product in development; some statements are forwarding looking and are subject to a variety of risks and uncertainties. BD Libertas™ Wearable Injector is a device component intended for drug-device combination products and not subject to FDA 510(k) clearance or separate EU CE mark certification. BD, the BD Logo and Libertas are trademarks of Becton, Dickinson and Company or its affiliates. © 2025 BD. All rights reserved.
REFERENCES
- Schneider A et al, “Understanding patient preferences for handheld autoinjectors versus wearable large-volume injectors”. Expert Opin Drug Deliv, 2023, Vol 20 (2), pp 273–283.
- Prašnikar M et al, “Novel strategies in systemic and local administration of therapeutic monoclonal antibodies”. Int J Pharm 2024, Vol 667(Pt A), art 124877.
- Li Z, Easton R, “Practical considerations in clinical strategy to support the development of injectable drug-device combination products for biologics”. MAbs, 2018, Vol 10 (1), pp 18–33.
- “Draft Guidance for Industry and FDA Staff: Purpose and Content of Use-Related Risk Analysis for Drugs, Biological Products, and Combination Products”. US FDA, Jul 2024.
- “Draft Guidance for Industry and FDA Staff: Human Factors Studies and Related Clinical Study Considerations in Combination Product Design and Development”, US FDA, Feb 2016.
- “Draft Guidance for Industry and FDA Staff: Content of a Complete Submission for Threshold Analyses and Human Factors Submission to Drug and Biologic Application”. US FDA, Sep 2018.
- “Draft Guidance for Industry and Food and Drug Administration Staff: Content of Human Factors Information in Medical Device Marketing Submissions”. US FDA, Dec 2022.
- “Skyrizi package insert”. Abbvie, Jun 2024.
- “Empaveli package insert”. Apellis Pharmaceuticals, Sep 2023.
- “Draft Guidance for Industry and FDA Staff: Applying Human Factors and Usability Engineering to Medical Devices”. US FDA, Feb 2016.