

To Issue 147
Citation: Allmendinger A, Adler M, Mahler H-C, “Integrated Drug Product Development and Manufacturing.” ONdrugDelivery, Issue 147 (May 2023), pp 44–50.
Andrea Allmendinger, Michael Adler and Hanns-Christian Mahler highlight the importance of integrated drug product development, considering aspects such as formulation, device, container closure system, manufacturing processes and usability, as well as considering the quality target product profile and clinical phase-appropriate technical development strategy.
“Drug substance manufacturing is followed by sterile drug product manufacturing, also referred to as fill-finish processing.”
The ultimate goal of the technical development and manufacture of parenteral products is to develop and supply safe, effective, patient-friendly and competitive medicines for patients. These sterile products must meet regulatory and compendial requirements, as well as related quality aspects, to support clinical studies and deliver for the market after product launch.
Drug substance development of biologics focuses on establishing cell-derived manufacturing processes, yielding purified, efficacious molecules with a certain impurity profile (process- and product-related impurities), however, sterile product manufacturing does not end here. Drug substance manufacturing is followed by sterile (typically aseptic) drug product manufacturing, also referred to as “fill-finish” processing, as an integral part of manufacturing operations. Fill-finish processing is associated with different complex, often underappreciated challenges to supply the final sterile drug product.
This article focuses on the interplay between drug product formulation, primary packaging and drug product manufacturing processes. It highlights how to approach and design the overall technical development to commercialise the final product and avoid lengthy and costly delays by anticipating failure modes and developing mitigation strategies.
TECHNICAL DEVELOPMENT ROAD MAP
State-of-the-art development of parenteral dosage forms comprises the development of formulation and manufacturing processes using appropriate primary packaging materials and possibly devices, and developing and using appropriate analytical methods according to quality-by-design (QbD) principles. QbD is a systematic approach to development that starts with predefined objectives and emphasises product and process understanding based on sound science and quality risk management as described in the ICH Q8 and Q9 guidelines.
“The content of a product’s TPP and QTPP are typically defined according to patient needs based on the benefit and potential risks to the user or patient population, but also according to market projections, competitive market advantage and regulatory and compliance expectations.”
Following QbD principles, technical product development starts with the end product in mind, which is summarised in the target product profile (TPP). The TPP typically describes the product in detail and includes aspects such as indication, patient population, treatment duration, delivery mode/route of administration, dosage form, regimen, efficacy, side effects and therapeutic modality. The quality target product profile (QTPP) links the TPP with expected quality and product aspects, and comprises dosage form, delivery systems, dosage strength, container closure system, route of administration, shelf life, storage temperature, purity and compendial compliance (such as sub-visible and visible particle levels), among other aspects.
The content of a product’s TPP and QTPP are typically defined according to patient needs based on the benefit and potential risks to the user or patient population, but also according to market projections, competitive market advantage and regulatory and compliance expectations. As an example, a subcutaneous (SC) application will have a higher user acceptance, and therefore probably higher market penetration, when developed for self-administration and supplied in a prefilled syringe, autoinjector or on-body device compared with a vial presentation due to a significantly higher ease of use. Another example is the higher acceptance for intravitreal injections if dosing frequencies are kept to a minimum.
CLINICAL PHASE-APPROPRIATE TECHNICAL DEVELOPMENT APPROACH
To support clinical and preclinical toxicological studies, a technical development strategy is typically designed by considering a clinical phase-appropriate approach (Figure 1). While, during early clinical and preclinical studies, a high degree of flexibility is required with respect to the applied dose and likely the route of administration, ideally, the final drug product formulation in the final primary container should be used for pivotal studies before product launch.
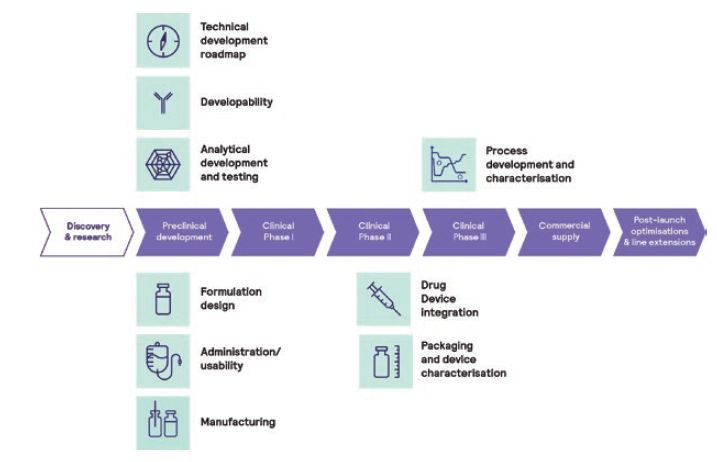
Figure 1: Phase-appropriate technical development approach.
“For biologics, an adequate formulation needs to be developed as early as possible to stabilise the protein and ensure adequate shelf life to support clinical studies.”
The therapeutic dose is typically unknown for entry into human (EIH) studies, due to only having limited pharmacodynamic and pharmacokinetic data available from pharmacological and toxicological studies, sometimes with limited applicability of in vitro or in vivo (animal) tests to the human situation. Therefore, the effective clinical dose needs to be defined in dose escalation studies in humans. In particular, the intended starting dose must be aligned between technical and clinical disciplines. As a result, the drug product configuration must be chosen anticipating changes to the dose regimen.
Vial configurations are a very flexible configuration if developed in combination with an appropriate formulation, allowing for a broad range of fill volumes and different administration options, such as intravenous (IV) or SC injection, dilution in infusion bags followed by infusion or injection via an injection pump. This can be useful, as several drug product strengths and configurations may have to be manufactured during clinical development in order to supply the changing demands and needs of clinical studies.
It is worth noting that each change in drug product configuration, such as a change in fill volume or container closure system (CCS), will require the submission of an amendment to the regulatory dossier. Changes are also associated with additional stability studies, and the impact and potential relevance of these changes on clinical studies needs to be technically assessed in detail and considered with respect to the development timelines and costs.
Many molecules entering the clinical development phase will not make it to market because of an unfavourable risk/benefit balance, such as a lack of efficacy, poor safety findings or an inferior clinical read-out compared with the expected standard of care. Therefore, it is desirable to opt for a lean and cost-conscious development and manufacturing approach until proof of concept (PoC) has been shown. However, reaching the market as fast as possible cannot compromise product quality at any point, with the highest priority being patient safety.
Especially in drug substance development, major changes may occur during technical development, requiring a fine balance between “speed” and “representativeness”. Product specifications as documented in the QTPP are typically linked to clinical exposure and variability during clinical testing.
For biologics, an adequate formulation needs to be developed as early as possible to stabilise the protein and ensure adequate shelf life to support clinical studies. However, little is known at this stage of development about product stability during manufacture and storage, as well as about the compatibility of the product with manufacturing and administration materials. Platform-based approaches to formulation and primary packaging are often recommended so that developers can build on existing knowledge and experience, which can – but does not have to – then be refined at a later stage of development.
Some companies prefer a lyophilised formulation over a liquid for EIH studies to maximise product stability. However, the higher manufacturing costs and lower end-user convenience of lyophilised formulations mean that it is likely that the drug developer will want to switch to a liquid formulation at a later point in development. Changing from a lyophilised to a liquid product also creates some specific technical challenges, as new stability studies for the liquid formulation are required and can be expected to show higher degrees of degradation compared with the lyophilised dosage form.
In such cases, the post-change product is expected to be less stable than the pre-change product. With a lack of clinical exposure to relevant impurities and degradants in early-stage clinical testing, this approach not only requires a significant effort to make the change in late-stage development, but also bears a risk of requiring further preclinical, or even clinical, studies to evaluate any potential safety liabilities of the post-change product.
SWITCHING FROM VIAL TO DRUG-DEVICE COMBINATION PRODUCT
The route of administration should be defined in the TPP/QTPP. For injectables, SC application, in combination with a ready-to-use injection device, is typically preferred over IV application, especially for chronic diseases, as well as increasingly for oncology treatments. This can provide benefits such as enabling at-home delivery, shorter administration, self-administration and flexibility for the user. However, the development of drug-device combination products is cost intensive and comes with additional technical risks, meaning that it is typically pursued during later clinical phases.
For EIH, a traditional vial configuration provides maximum flexibility to react to the needs of the clinical dosing schedule, especially as the dose range is unknown for Phase I studies. For known and established vial configurations, the technical risks are rather low, in large part due to the pre-existing knowledge about their use. In early-stage development, leveraging extensive formulation and process development know-how and experience can provide a significant advantage towards defining and making the right experiments using the right methods, which can provide a boost in quality and time compared with generating hundreds of formulation combinations or data points from a blank slate. Another consideration is that fewer stability data at intended storage temperatures are required for regulatory submission for early clinical phases.
The switch from a vial format to the drug-device combination product is associated with numerous technical risks; comparability needs to be demonstrated between the two dosage forms, including comparative stability studies. However, many technical and analytical endpoints can be expected to be different when assessing the product in the vial versus in a syringe. Manufacturing vial and prefilled syringe or cartridge presentations on the same filling line can avoid technical transfers and thereby minimise technical and comparability risks, as well as costs and delays to project timelines.
Clinical bridging when switching from a vial format to a combination product is ideally performed prior to the start of pivotal studies to minimise any risks associated with the switch. Switching configurations during the pivotal phase is still possible when pursuing accelerated development options; however, additional clinical study arms may be needed.
Switching the route of administration during clinical development, such as from IV to SC, in addition to introducing a combination product, adds further complexity to both the technical and clinical development road maps, requiring extra clinical bridging studies, such as bioavailability and safety studies, as well as technical comparability studies if the API concentration or formulation is adapted.
“The switch from a vial format to the drug-device combination product is associated with numerous technical risks; comparability needs to be demonstrated between the two dosage forms, including comparative stability studies.”
CCS AND DEVICE SELECTION – SELECTING THE RIGHT PARTNERS
Selection of the appropriate CCS and injection device for a formulation, such as a prefilled syringe, autoinjector, pen or on-body injection device, depends on the target patient population and indication, intended use or user preference, to name only a few potential considerations. Besides usability aspects, technical challenges during drug product development and manufacturing must be evaluated when selecting the appropriate CCS and device. Technical challenges comprise product compatibility with the primary packaging components, product stability, compliance with compendial tests (such as particulates) and functionality.
Functionality and usability are interlinked with the product formulation, particularly so with its viscosity and its visco-elastic behaviour, as well as with manufacturability and choice of unit operations and specific process set-up. Of course, the design of the appropriate needle, including its size, shape, type and supplier, is key. Novel CCSs should be assessed for container closure integrity in detail, using the most sensitive methods, such as helium leakage, and not relying on crude, probabilistic tests, such as dye ingress testing.
The criticality of the entire CCS’s material attributes should be studied in detail, considering the respective interplay between parts, including dimensional variances. For example, the variability of a plunger stopper’s elasticity in a syringe barrel may significantly impact the quality of the stopper setting during fill-finish operations.
The CCS should initially be chosen to cover a range of fill volumes and viscosities and provide a reproducible injection time per dose strength to allow some flexibility during product development. Selection of the appropriate device and CCS must also consider the pros, cons and associated risks of selecting a new device versus an established CCS.
From a technical perspective, an integrated and holistic development approach, including formulation sciences, manufacturing operations and primary packaging and device component performance and quality testing, is recommended (Figure 2). Selecting the right partners according to their capabilities is essential for transferring product knowledge and facilitating troubleshooting activities. Even better, the groups developing the formulation, choosing the primary packaging and device, setting up and defining the manufacturing process, handling the fill-finish facility and managing quality control and assurance should be within the same company and entity.

Figure 2: Considerations for integrated drug product development.
Strategy and project timelines must be synchronised between all teams and specifications and test methods need to be aligned to ensure lean technical transfer from development to manufacturing. Additionally, troubleshooting activities can be approached holistically to save time, cost and resources, as well as, most importantly, to avoid errors. In summary, it is recommended to select a partner for manufacturing with very strong development capabilities and expertise.
THE ART AND SCIENCE OF DRUG PRODUCT DEVELOPMENT
Previously, formulation development has sometimes been considered as a by-product of drug substance development, with focus on a few biophysical parameters, such as unfolding or melting temperature of the molecule. However, appropriate formulation development based on expert knowledge using relevant analytical methods and endpoints can resolve many challenges along the supply chain, as a drug product must be manufacturable, shippable, stable during intended or accelerated storage and easy and safe to administer, as well as fulfilling compendial requirements and related quality aspects.
To succeed, a drug product formulation should be developed as soon as possible – ideally, before entering into preclinical and clinical studies, considering all product aspects, such as the target product profile, the intended manufacturing process, its primary packaging and, last but not least, usability by the patient and healthcare professionals.
“The manufacturing processes for early-phase clinical supplies should make use of prior knowledge when setting up the unit operations and when defining target process parameters by using established product-independent process parameters, such as capping pressure, or by using platform CCSs.”
Early-Stage Development and Manufacturing
The extent and timelines for early-stage formulation development studies generally depend on the molecule. Whilst platform formulations are well established for monoclonal antibodies and antibody fragments, formulation screening is usually recommended for complex molecules, which typically include pH/buffer and excipient screens. Excipients must be safe and non-immunogenic within the dose ranges used, approved for parenteral use and available in a parenteral grade. Information from developability assessments and forced degradation testing, such as by pH, oxidation, light stress, isoelectric point or hydrophobicity, can be helpful to guide the design of the formulation screens.
To define the target concentration for high-concentration protein formulations, it is important to study the relationship between viscosity and protein concentration, the visco-elastic behaviour and the potential need for viscosity-reducing agents prior to the excipient screen. The target protein concentration for the excipient screen is then based on the outcome of the viscosity assessment. It is typically a compromise between injectability, manufacturability and acceptable injection volume to achieve the desired dose.
As the formulation components can interact with the components of the CCS, formulation development studies should be performed using representative CCSs, which are typically vial configurations for early-phase clinical studies. Examples of such interactions include the precipitation of inorganic material, which results from leaching of bivalent ions with formulation components, and the occurrence of glass surface defects over time, such as delamination.
Potential protein degradation pathways include chemical degradation, such as deamidation, and physical degradation, such as soluble aggregation or the formation of proteinaceous sub-visible or visible particles. Therefore, it is important to use a broad, relevant analytical method panel, including detection and counting of visible and sub-visible particles beside chromatographic and electrophoretic methods for protein purity.
Biophysical methods, such as thermal stability by differential scanning fluorimetry or colloidal stability by dynamic light scattering, have not been proven to be fully predictive for stability yet. Therefore, these methods can support but not replace the need for short-term stability studies and the application of a broad stability-indicating analytical method panel. A forced degradation study should be performed prior to formulation development to establish stability-indicating methods for protein quality. Potency methods are rarely suitable for formulation development studies, especially in early-stage trials, due to their inherent variability, meaning that they may not capture differences in the stability of different formulations.
The manufacturing processes for early-phase clinical supplies should make use of prior knowledge when setting up the unit operations and when defining target process parameters by using established product-independent process parameters, such as capping pressure, or by using platform CCSs. This eliminates the need for extensive process development studies prior to process implementation, with the added advantage of shortening timelines and saving API and cost. Platform manufacturing processes are generally applicable to platform molecules for which a sound formulation development has been performed, including short-term stability, freeze-thaw stability and shaking stress stability studies. The data from these studies aims to look for potential liabilities, which may impact manufacturability.
For more challenging molecules and formulations, such as high-viscosity formulations, process development studies should look at manufacturability with regard to freeze-thaw stability, compatibility with filter and other process materials, filter binding of API and surfactant, filling pump compatibility and temperature, ambient light and oxidation sensitivity. It is recommended to perform these studies using appropriate small-scale models in laboratories, rather than trying to perform development studies in costly GMP facilities.
“For the final manufacturing process, a thorough risk assessment should be performed according to ICH Q9 for example, a failure modes and effects analysis to identify potential critical process parameters.”
Late-Stage Development and Manufacturing – Before Start of Pivotal Phase
Late-stage development starts prior to pivotal clinical trials with formulation optimisation studies that define a formulation in its final CCS in a format suitable for commercialisation. The aim is to optimise stability and thus maximise shelf life by adjusting formulation parameters based on existing long-term stability data of the early-stage formulation. In many cases, the API concentration and dosage strength may also be adjusted during formulation optimisation based on information from clinical dose-finding studies.
The final CCS and device is selected with consideration of the intended route of administration, injection volume, solution viscosity and manufacturability, amongst other factors. Injectability is impacted by solution viscosity, needle gauge and the properties of the primary packaging components according to Hagen-Poiseuille’s law. Additionally, siliconisation of the device or stopper hardness may impact injection forces.
Especially when considering ready-to-use containers and closures, it is important to understand how well particle contamination is prevented and controlled by the manufacturer, as particles cannot be removed during the manufacturing process of the drug product. Prior to the introduction of a new CCS, container closure system qualification needs to be performed to ensure container closure integrity, which maintains sterility of the drug product. Additionally, it is essential to assess the compatibility of the formulation with the primary packaging components prior to switching, for example, from a vial used for early-phase clinical trials to a prefilled syringe for late-phase clinical trials or post-approval lifecycle management.
As prefilled syringes and cartridges are far more complex and have more materials in contact with the drug product than a vial, incompatibilities resulting in protein adsorption, protein degradation and formation of proteinaceous sub-visible or visible particles can occur. Prefilled syringes require lubrication to ensure functionality during the product’s shelf life. Traditionally, glass syringes use silicone for that purpose. Siliconised primary packaging components typically release silicon oil droplets into the formulation, resulting in an increase in sub-visible particles.
It is important to characterise the sub-visible particle population with methodologies such as flow-imaging microscopy. This enables differentiation of silicon oil droplets from proteinaceous particles, which is valuable as some protein formulations are sensitive to silicon oil, resulting in proteinaceous particles. Such studies are also helpful when changing from a vial to a delivery device, given that this results in higher sub-visible particle counts are expected, yet, typically, not of any clinical significance.
There are further challenges relating to the use of syringes, as residual tungsten in glass prefilled syringes has been reported to lead to protein aggregation and protein oxidation. Radicals in gamma irradiated cyclo-olefin polymer syringes may also result in protein aggregation. Furthermore, cyclo-olefin polymers are permeable to oxygen, which can lead to oxidation of the API or other formulation components. In cases where oxidation is a critical quality attribute, such as with therapeutic proteins that contain a methionine in their binding regions, oxidation events can significantly diminish product efficacy.
In general, incompatibilities can result in a change in product quality leading to underdosing, a shorter shelf life or even requiring a safety assessment. Thus, it is prudent to plan compatibility studies with the selected final CCS and device well ahead of introducing it in late-phase clinical studies so that alternatives can be identified in case of incompatibilities without impacting the overall project timelines.
During development of a prefilled syringe with a staked-in needle, the risk of needle clogging needs to be assessed. Needle clogging can occur due to water vapour transmission from product solution in the needle through the rigid needle shield. Needle clogging is especially critical for high-concentration protein formulations as the solution in the needle can solidify. The impact of a solidified plug is partial or no delivery of the dose and a failure in design verification testing. A careful selection of the prefilled syringe components can mitigate the risk of needle clogging.
Functionality testing of devices is required during formulation development to assess if, and to what extent, break-loose and glide forces change dependent on storage condition and time. Injection forces testing must also address the user capabilities as evaluated in human factors studies. In siliconised prefilled syringes and cartridges, the release of silicon oil from the barrel dependent on storage time might result in an increase in break-loose and glide forces or an increase of injection time for autoinjectors. This impact can result in a failure in design verification and lead to out-of-specification results for stability.
Ideally, the manufacturing site for early-phase clinical supplies can support the transition from the early-phase CCS, such as a vial, to the late-stage and commercial CCS, such as a prefilled syringe or cartridge. The manufacturing process can essentially remain unchanged apart from the filling operation, which saves time and cost for a technical transfer, as well as minimising the risk of comparability failures. Filling parameters need to be optimised and tubing and needle diameter need to be adequately chosen to ensure fill weight accuracy in manufacturing. Furthermore, the stoppering process parameters, for example, the vacuum setting in the case of vacuum stoppering, might need to be determined depending on prior knowledge and formulation characteristics.
The implementation of filling processes for innovative CCSs, such as prefilled syringes for intravitreal injection and cartridges as well as for formulations with challenging formulation properties, may require specific expertise and know-how. The challenges involved with novel CCSs are manifold, such as fill weight accuracy for low fill volumes or bubble-free stopper setting.
Late-Stage Development and Manufacturing – During Pivotal Phase
During pivotal clinical studies, the robustness of the formulation should be tested to assess the impact of formulation parameters on product stability over its shelf life, such as protein and surfactant content or pH. During routine manufacture, various parameters of the formulation are expected to vary within the predefined ranges, such as pH or concentration, therefore, it must be ensured that the product quality remains acceptable. Depending on the results of such a formulation robustness study, the drug product release specification can either be supported by the stability data or it may need to be tightened to ensure quality throughout the intended shelf life. Furthermore, container and closures extractables and leachables studies should be initiated during pivotal studies, in conjunction with ICH stability studies, which need to be submitted in the biologics license application or market authorisation application.
For the final manufacturing process, a thorough risk assessment should be performed according to ICH Q9 – for example, a failure modes and effects analysis – to identify potential critical process parameters. As a result, process characterisation studies should be performed to evaluate the impact of the manufacturing unit operation, as well as any associated potential critical process parameters and their ranges, including time out of refrigeration, ambient light exposure and extractables and leachables of product contact materials, on critical quality attributes and process performance.
These studies should identify critical and non-critical process parameters and enable the definition of target and acceptable process parameter ranges. Based on the results of process characterisation studies, necessary and meaningful in-process controls and respective acceptance criteria or alert limits can be defined, assummarised in the overall control strategy. After successful process characterisation, the risk assessment is usually updated and the process performance qualification campaign can be executed as prerequisite for a biologics licence application or market authorisation application submission.
To enable an impact assessment on product quality after temperature deviations during transport, it is recommended to perform a temperature excursion study. High and low temperature excursions are simulated and their impact on product stability is tested. Furthermore, the impact of transportation on product quality can be assessed by transport simulation studies, which simulate mechanical stress during air and ground transportation in a lab-scale set-up. The impact of reduced pressure during air transportation on potential stopper movement in prefilled syringes and cartridges, which might impair sterility, can be tested at lab scale with an appropriate vacuum chamber. It is key to appropriately define representative worst-case samples when performing such studies, for example, the air bubble size will typically be relevant for prefilled syringes.
CONCLUSION
Given the many pitfalls and challenges covered in this article, it is obvious that the development and manufacturing of a drug-device combination product goes far beyond the identification of a stable, high-concentration protein formulation or the ability to successfully fill a GMP batch of sterile product. It requires the expertise and experience to select a formulation, container closure system and suitable device, as well as to define appropriate manufacturing processes and product use, considering all possible interactions and failure modes.



