


To Issue 128
Citation: Shortt N, “Software as a Medical Device: from a Human Factors Perspective”. ONdrugDelivery, Issue 128 (Dec 2021), pp 22–25.
Natalie Shortt discusses the considerations and steps to take when planning usability engineering efforts for software as a medical device.
“To determine whether your software is SaMD, you need to craft an intended use statement.”
A quick search online will show that guidance relating to “software as a medical device” (SaMD) is sparse, particularly when it comes to applying usability engineering to software. When a client approaches with what might be SaMD, it is important to run through a series of questions to determine exactly what will be required for that piece of software.
The client might be a traditional medical device manufacturer or a pharmaceutical company that has decided to develop a connected technology to accompany their treatment pathway. However, occasionally there are software developers that have found their innovative software is encroaching on medical territory, and so are bound by medical regulations. In any situation, the same initial stepping stones help to understand what activities are required. This article maps out a range of considerations and steps, from a human factors perspective, that enable you to plan your usability engineering efforts accordingly.
CONFIRMING IT IS SAMD
First and foremost, it is necessary to determine which of the following the software is:
- Software that is not a medical device
- Software that is integrated into a medical device (Figure 1)
- Software, independent of any hardware, that contributes towards medical care.
The last of these is SaMD. The International Medical Device Regulators Forum (IMDRF), a voluntary group of medical device regulators from around the world, has developed guidance that can help to determine what the software should be classed as.
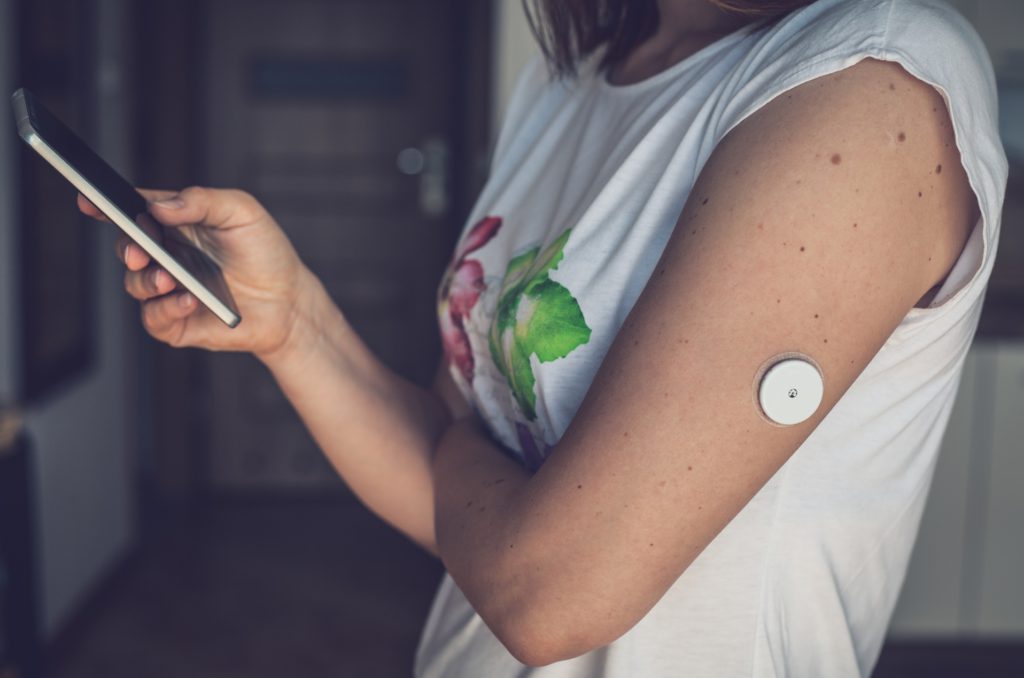
Figure 1: A woman using her phone to review the data being collected by her continuous glucose monitor. The monitor does require the mobile app to function, but the mobile app may influence the patient’s decision making in monitoring their glycaemic index. Therefore, this could be considered software integrated into a medical device.
“The intended use statement can be used to determine if the software meets the definition of SaMD or if it is an integrated part of the overall medical device.”
The IMDRF defines SaMD as “software intended to be used for one or more medical purposes that performs these purposes without being part of a hardware medical device”,1 whereby “without being a part of” means software that is not necessary for a hardware medical device to achieve its intended medical purpose. To this end, to determine whether your software is SaMD, you need to craft an intended use statement. This will help to understand if the intended use is integral to the overall use of a medical device. If this intended use statement indicates that the software is standalone from the device, the software is more likely to be SaMD.
The IMDRF suggests an intended use statement should consist of three major points:
- A clear and strong statement about intended use, outlining whether the device is used to: treat or diagnose, drive clinical management, inform clinical management.
- The state of the healthcare situation or condition, either: critical, serious, non-serious.
- A description of the software’s core functionality, identifying critical features essential to the intended significance of the information the software provides.
As an illustrative example of an intended use statement, consider “a piece of software that provides information regarding insulin uptake in people newly diagnosed with Type 1 diabetes, so that they can observe whether they are calculating their insulin correctly per meal. The software reviews data pertaining to insulin levels pre-meals, post-meals and post-administration of an insulin dose, and provides an outline of any discrepancies in the maths”.
The intended use statement can be used to determine if the software meets the definition of SaMD or if it is an integrated part of the overall medical device. In both cases, the device is subject to usability engineering, but what that will look like may change according to the overall objective. Additionally, for software to be considered a medical device, it must meet the criteria for a medical purpose. The IMDRF suggests taking the definition of the term “medical device”2 into account. This definition will vary, depending on the market. In the EU, it is best to review the definition of a medical device as per the Medical Device Regulation, whilst in the US it would make sense to review the formal terms provided by the US FDA.
CLASSIFYING THE LEVEL OF RISK
Once you have discussed the intended use of the software and feel confident that it is SaMD (or at least a part of a medical device), there is a framework for determining what level of risk may be associated with the SaMD.
Within the EU, at least, SaMD is automatically considered Class IIa (generally a low-medium risk device) by default, so initial assumptions should be that manufacturers will at least have to perform some usability engineering, develop usability documentation and demonstrate compliance with a usability engineering file. To get a better idea of exactly what level of effort is required, a systematic process can be followed.
Start with the IMDRF guidance N24, “Possible Framework for Risk Categorization”2 – this will give an indication of the level of risk that may be associated with the device, and therefore what level of usability engineering should be applied during development. This categorisation is independent from regulatory classification and the two should not be confused.
To get an idea of what level of risk the SaMD is, return to the intended use statement from before. Risk associated with SaMD is considered through two variables:
- Significance of information provided by SaMD to healthcare decision
- State of healthcare situation or critical condition. IMDRF N24 lays this out as a table (Table 1).
| State of healthcare situation or condition | Significance of information provided by SaMD to healthcare decision | ||
| Treats or diagnoses | Drives clinical management | Informs clinical management | |
| Critical | IV | III | II |
| Serious | III | II | I |
| Non-serious | II | I | I |
Table 1: SaMD risk classification (IMDRF N24).
From the intended use statement, you can reason the type of information that the SaMD will provide. You can also infer the state of the healthcare situation or condition (e.g. the state the patient is in when receiving care with the SaMD). The combination of these two variables gives an indication of how much risk may be associated with normal use, with I being low risk and IV being high risk. In the case of the diabetes management example, we know it provides information relating to a serious healthcare condition. Therefore, it would be somewhere between low and low medium risk. Although these categories are not regulatory categories, the level of risk can be used to gauge what formal class the SaMD should be – whether to stick with the automatic Class IIa or not.
“One of the immediate difficulties is developing a test plan for something that is frequently changed.”
MANAGING THE DOCUMENTATION
At this point, you should be confident about whether or not your software is SaMD and what pathway to consider. Let’s assume the device is Class IIb – perhaps the SaMD is intended to drive clinical management in a serious healthcare situation – there will be an expectation to conduct full usability engineering and demonstrate compliance with a usability engineering file.
For hardware devices, this is already quite a task – but changes are implemented slowly, which means documents are only updated periodically, perhaps at the conclusion of each milestone or once or twice a year. For SaMD, this is not the case. Software benefits from being easily edited or updated based on design decisions – you can enter a meeting with one iteration and make some design changes within the meeting, go to another meeting with that iteration and then make some more design changes within that meeting! This is great for refining the device, but is problematic for usability engineering because these changes should technically be documented. At the rate of software development, someone would have to update usability documentation on a daily basis, which is impractical.
To reduce the impact quality management can have on the creative design process for software, manufacturers can benefit from setting some internal rules or processes that trigger the need to update documentation. This could be included in a “usability engineering plan”, a document that contributes to demonstrating compliance. Within the plan, specify what magnitude a design change needs to be to trigger a document review, then ensure that all team members who have the capacity to make interface changes understand the triggers.
Additionally, the plan should include a periodic timeframe for updating documents, with an agreement on how versions of the software are managed. For example, this plan could specifically outline that documents will be reviewed every three months. On the xth day of that month, the most up-to-date version of the software will be considered the current version and all documentation will be updated with that iteration. This is a structured approach that implements the methodological practices that are typical to medical device risk management, whilst trying to maintain the creative processes common in software development.
CONDUCTING USABILITY STUDIES
Similarly, one of the immediate difficulties is developing a test plan for something that is frequently changed. In usability studies, it is typical to define the use flow within the protocol and describe what is being assessed at each point in the use flow.
Furthermore, these documents can take weeks or months to develop. However, in software development a lot can change in the space of a few weeks, let alone a few months, so the protocol author needs to take this into account. The author needs to develop a protocol that is flexible to meet the needs of prospective study changes. The author may therefore need to be involved in various meetings to gain a greater understanding of the plans for the SaMD over the following weeks. This will give them some insight for developing a protocol with moving goalposts.
Participant interpretation of software has been shown to be largely different from hardware and this must be taken into account when developing the study script as well.
Participant Interpretation of Software Versus Hardware
Users interact with software completely differently from hardware. Imagine you are given a comprehensive blueprint of a house. Quite easily, you could see all the rooms and understand the design of the house. Now imagine you were placed at the front door of a house you’ve never been into and asked to find the bathroom on your first attempt. Would you be able to find the bathroom on your first attempt? Maybe not!
In late-stage usability studies, participants are traditionally given one attempt to get something correct, which makes sense if you have the blueprints (in this case, a physical product and all components) but not so much if you have no idea what’s inside – looking at the log-in screen for software does not indicate the internal functionality that is waiting. So, how does one comply with usability engineering requirements and also give users a fair chance at using the product? One answer is to build in some exploratory time with the app to give participants an opportunity to become familiar with the “inside of the house”. Ensure that you build this into the most realistic portion of the session – typically this is once the user has logged in for the first time.
Also, key to late-stage usability testing are the success and failure criteria for each isolated task. Hardware usually has a very limited number of logical actions, whilst software has more, and the list grows greater with the more features that are included in the software. For example, to use a pen injector a user must remove the cap. They can either remove the cap correctly, not remove it at all or remove it in such a way that it damages the device. This is a limited scope of logical actions. However, with software a user could click the right option or one of several incorrect options. Once they have made a choice, they are faced with the opportunity to click the right option again or several incorrect options. Recording all the missteps generates a lot of data, so success and failure criteria need tobe designed to encompass an entire activity, rather than the sub-tasks that contribute to the overall activity. This option is only appropriate when it reflects the context of use – if there are sub-tasks that can have effects later in use, these tasks would need to be assessed independently.
SUMMARY
So, to drive effective usability engineering for software, make sure to determine early on whether it is indeed software in a medical device, software as a medical device or simply software, by developing an intended use statement. Once you have this, consider possible risk categories for software and use this to help understand what class of device you are developing. Finally, use this knowledge to tailor the usability engineering effort to meet the needs of the software development process, remembering to make sure that your study protocols are tailored to using software rather than hardware.
REFERENCES
- “Software as a Medical Device (SaMD): Key Definitions”. IMDRF, Dec 9, 2013.
- “Software as a Medical Device: Possible Framework for Risk Categorisation and Corresponding Considerations”. IMDRF, Sep 18, 2014.
