To Issue 130
Citation: Ritter A, Birkhoff M, “An Eye on the Future of Preservative-Free Drops”. ONdrugDelivery, Issue 130 (Mar 2022), pp 20–26.
Alexandra Ritter and Matthias Birkhoff discuss the importance of the regulatory support the company offers to its customers for preservative-free ophthalmic platforms in the face of increasingly stringent regulatory requirements from both the EU EMA and US FDA, and describe new opportunities and growing demand for a wider range of multidose preservative-free ophthalmic drug-device combinations, including for indications such as dry-eye and glaucoma.
“Patients almost universally prefer to receive eye medication via topical drops, whenever the format is available.”
As the global population continues to live longer, vision care products are increasingly important, for both general vision care as well as quality of life, for those with various ocular disorders. Global sales of ophthalmic drugs represented approximately US$28.4 billion (£21.2 billion) in annual sales (2020),1 with sales expected to grow to nearly $48 billion by 2030. Many new drugs are being developed and approved to treat ocular conditions such as dry eye syndrome, glaucoma, red eye and conjunctivitis (Figure 1). Furthermore, retinal disorders such as dry and wet forms of age-related macular degeneration (AMD) or diabetic retinopathy demonstrate clearly that ophthalmic treatments are a growing market segment.
Figure 1: Ageing populations are one driver of the growing need for ophthalmic treatments.
Most medications for treating retinal diseases and conditions are only available as invasive ocular injections. Typically, retinal diseases are chronic conditions requiring ongoing drug administration and, with many patients receiving regular injections into their eyes, this can result in reduced patient compliance and poorer patient outcomes overall.
Patients almost universally prefer to receive eye medication via topical drops, whenever the format is available, as opposed to having to go to a healthcare professional to receive repeated painful injections to the eye.2 For a number of ocular diseases, eye drops provide a convenient and comfortable way to self-administer ocular medication.
So, what are the challenges to administering a wider range of medications via topical eye drops?
“Studies have shown that long-term preservative use can lead to ocular hyperaemia, dotted keratitis or toxic keratopathy.”
PRESERVATIVES
For one, multidose eye-drop formulations typically include preservatives to avoid micro-organisms contaminating the sterile eye-drop solution. This can occur when a normal eye-drop device is exposed to these contaminants by coming into contact with the eye or through other sources in the environment. The most common chemical preservative used in nasal and eye-drop formulations is benzalkonium chloride, which serves as a detergent, antiseptic, disinfectant, fungicide and bactericide.3 Such preservatives have been shown to lead to undesirable side effects, particularly from long-term use, ranging from temporary irritation and discomfort to even more complex issues such as dissolution of the lachrymal film, apoptosis (cell death) and inflammation.4,5 Studies have shown that long-term preservative use can lead to ocular hyperaemia, dotted keratitis or toxic keratopathy.
Another approach to maintaining eye-drop sterility is to apply silver ions or surface coatings to ophthalmic devices. These can provide antibacterial and antifungal protection of the formulation but can also present their own challenges as they may become less effective over time as the silver materials are consumed6 or, more concerningly, could have accumulated health effects on the user.7
THE PRESERVATIVE-FREE OPHTHALMIC DEVICE SOLUTION – APTAR PHARMA’S OSD
One way to enjoy the benefits of ophthalmic eye-drop delivery while avoiding the risks of using preservatives is to use a multi-use eye-drop device designed specifically to support preservative-free formulations. This requires a specialised ophthalmic eye-drop device that maintains the sterility of the liquid formulation, even with repeated use of the device throughout the treatment period.
Aptar Pharma manufactures the only ophthalmic device component that has been reviewed by the US FDA as part of the approval of a preservative-free multidose (PFMD) ophthalmic drug-device combination product. Aptar Pharma’s Ophthalmic Squeeze Dispenser (OSD) uses a patented, purely mechanical Tip-Seal technology (Figure 2A) to maintain formulation sterility within the device. It also has a metal-free formulation pathway that avoids oxidative reactions between the formulation and metals.
This unique OSD device works as follows:
- As the bottle is squeezed, the pressure inside the Tip-Seal rises until the spring-controlled sealing membrane opens and releases a single drop (Figure 2B and C).
- The Tip-Seal then reseals the tip orifice, blocking potential contaminants from entering the tip.
- To equilibrate the created pressure difference, air flows into the squeeze bottle through a microbiological filter, preventing contamination of the formulation (Figure 2D).
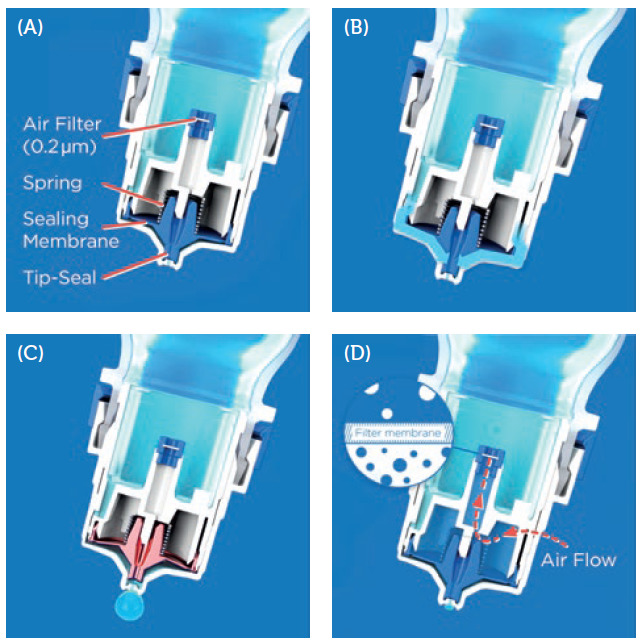
Figure 2: The patented Tip-Seal mechanism of the OSD. (A) Aptar Pharma’s OSD Tip-Seal technology with a sealing membrane, spring mechanism and microbiological filter, protecting the formulation from any bacterial ingress via inflowing air. (B) By squeezing the bottle, the pressure inside the system rises, allowing the formulation to flow into the delivery channels. (C) The pressure rises until the sealing membrane of the Tip-Seal opens to release the drop. (D) After the drop has been released, the Tip-Seal closes again and any incoming air is filtered sterile through the 0.2 μm filter membrane.
“The ideal solution for administering preservative-free ophthalmic products is a multidose device that does not use any additives or preservatives.”
Due to its innovative design features, Aptar Pharma’s OSD device allows consumers and patients to deliver a variety of preservative-free ophthalmic eye-drop formulations – solutions, emulsions or suspensions. From a technical perspective, the OSD is a viable solution to the preservative problem for both consumer products and prescription eye-care medications – and offers a proven track record of more than 300 market references worldwide.
But what about the regulatory environment? How do regulatory authorities view a device-driven preservative-free solution for ophthalmic eye-drop products, and what regulations must the applicant comply with?
DRUG-DEVICE COMBINATION PRODUCT REGULATORY REQUIREMENTS
Leading regulatory authorities have been supportive of the drive to reduce or eliminate the use of side-effect-inducing preservatives for ophthalmic products. The ideal solution for administering preservative-free ophthalmic products is a multidose device that does not use any additives or preservatives. Aptar Pharma’s OSD technology platform offers a completely mechanical solution to meet these objectives. Products composed of both a device component (here, the OSD) and a drug product are viewed differently by regulatory authorities.
The FDA8 and the European Medicines Agency (EMA)9 have both continued to expand and clarify the requirements for these types of drug-device combination products (Figure 3). Aptar Pharma’s OSD device has already been accepted by regulatory authorities worldwide, including the FDA, Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) Germany, Agence Nationale de Sécurité du Médicament et des Produits de Santé (ANSM) in France and the National Medical Products Administration (NMPA) China. With new, even stricter regulations around sterile product manufacturing and validation, such as the Annex I of the Medical Device Regulation (MDR) 2017/745, which now applies to drug-device combination products in the EU, the path for ophthalmic eye-drop combination products has changed.10

Figure 3: Regulations for combination products, such as ophthalmic eye-drop combination products, have evolved for the US and Europe.
FDA Regulations
Under FDA regulations, therapeutic and diagnostic products that combine a drug or a biological product and a device are defined as combination products.11 The division of the FDA that takes the lead on the evaluation and approval of the combination product is determined by the primary mode of action (PMoA) of the product. Other FDA divisions are selected for other secondary, yet relevant, aspects of the combination product.
The Genus decision issued on April 16, 202112 by the US Court of Appeals DC Circuit, held that articles that meet requirements of Section 201(h) of the FD&C act must be regulated as devices and not drugs. Taking this decision into consideration, eye cups, eye droppers and ophthalmic dispensers that have been regulated as drugs when packaged with a drug according to 21 CFR 200.50(c) now meet the “device” definition. The FDA might begin regulating ophthalmic drug-led combination products where the drug is PMoA with the Center for Drug Evaluation and Research (CDER) as the primary regulatory branch overseeing these products. As a result, such combination products would also be subject to 21 CFR Part 4, including cGMPs and post-marketing safety reporting requirements.
If the combination product’s PMoA is a drug (not a device) then the combination product’s submission may follow the condensed 21 CFR 4.4(b) filing.13 This offers the benefit of requiring less information for the different constituent parts of the combination device, as outlined in option 2 below. For multidose (including preservative-free) eye-drop combination products, a drug manufacturer must now demonstrate compliance with cGMP regulations for each of the constituent parts (drug and device). There are two ways to achieve this requirement:
- Full approach – demonstrate compliance with all cGMP regulations applicable to the device and drug (full application for each constituent).
- Streamlined approach – with a condensed 21 CFR 4.4(b) filing for combination products, the drug manufacturer must demonstrate compliance with both the drug cGMPs (21 CFR parts 210 & 211) AND also demonstrate compliance with the following provisions of the device quality system (QS) regulation 21 CFR part 820:
a. 21 CFR 820.20 Management responsibility
b. 21 CFR 820.30 Design controls
c. 21 CFR 820.50 Purchasing controls
d. 21 CFR 820.100 Corrective and preventive action.
Most likely, design controls are the most significant challenge for a drug manufacturer. Design controls, as defined in 21 CFR 820.30, are a systematic process to ensure that the product design meets the user needs, intended use and regulatory requirements. This results in a design history file, which has to be maintained over the product lifecycle.
EU Regulations
In the EU, “combination products” are not recognised by the same naming convention as would be applied by the FDA. However, they are still regulated based on their PMoA, either as a medicinal product or medical device. A drug product that contains a medical device as an integral part can be submitted to regulators as either a medicinal product for drugs or biologics – or as an advanced therapy medicinal product for cell therapies. Multidose eye-drop combination products typically fall under the medicinal product category.14
In May 2021, the EMA launched a new regulatory framework applying to medicinal products when used in combination with a medical device. The EMA has also published a final guideline on quality documentation requirements for medicinal products when used with a medical device15 that took effect on January 1, 2022. Applicable eye-care products, such as those using Aptar Pharma’s OSD, have to meet expanded requirements, including elements such as Annex 1 of the MDR 2017/745.16 Most ophthalmic combination products would be reviewed as a medicinal product (drug) under Directive 2001(83/EC) or Regulation (EC) No 726/2004 and, in addition, would need to comply with the General Safety and Performance Requirements (GSPR) from MDR 2017/745, Annex I, in order to establish the safety and performance of the device, as well as the manufacturing requirements for sterile manufacturing.
If the product is to be marketed as a sterile “medicinal product” with an integral medical device, it would require the drug manufacturer to have a notified body opinion (NBOp). The NB would assess and confirm if the device meets the GSPR and would issue an NBOp, which must be submitted to the EMA in the MAA.
The drug manufacturer would need to provide comprehensive information to the NB to support the conformity assessment, such as data on risk management, safety, testing of performance, functionality and compatibility (accuracy of dosage, performance of device), usability, sterilisation validation and microbiology.11
RAISING THE BAR ON OPHTHALMIC EYE-DROP DEVICE COMBINATIONS
The outcome of these changes is that product owners of eye-drop combination products have to adapt to meet more comprehensive filing requirements that are focused on the device part. Partnering with a device manufacturer with proven experience of supporting applicants through the quality, regulatory and validation requirements, with both data and services, should be a critical consideration in every selection process.
APTAR PHARMA’S OSD DEVICE – REGULATORY SUPPORT
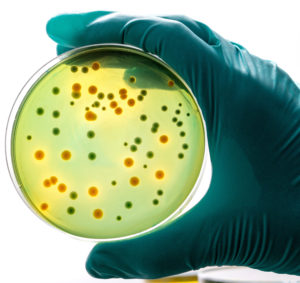
Figure 4: Aptar Pharma’s Tip-Seal Integrity Test TSIT 2.0 helps to define testing standards for PFMD devices.
Having a well-designed and manufactured eye-drop device alone is not enough to provide a de-risked and confidence-inspiring pathway to regulatory approval for your drug-device combination product.
Aptar Pharma did not stop at just designing and producing its innovative OSD system to meet market needs. It continues to develop data, support and services that help customers more easily navigate through the changing regulatory environment. With over 300 market references and millions of devices on the market, Aptar Pharma brings specialised experience with preservative-free eye-drop devices to every OSD project. For example, Aptar Pharma successfully supported the filing and FDA approval of a product for an ocular indication. The process benefited from the continued technical and regulatory support that Aptar Pharma was able to provide to the customer, including from its portfolio of stage-specific development packages. Aptar Pharma has enjoyed similar success in supporting product approvals with regulatory authorities in a number of markets around the world. Aptar Pharma has developed data packages around the form and validation of its Tip-Seal technology. It also offers a range of analytical testing and services to support the validation of the OSD devices. For example, Aptar Pharma’s Tip-Seal Integrity Test (TSIT) 2.017 challenges the mechanical tip closure system in worst-case scenarios against a number of bacteria and contaminants, including Pseudomonas aeruginosa (ATCC 9027), Staphylococcus aureus (ATCC 6538) and Candida albicans (ATCC 10231) (Figure 4).
These proprietary tests were designed to generate data supporting the integrity of the PFMD device suitable for submission to regulators while helping to define the testing standards for Aptar Pharma’s customers. This is just another way that Aptar Pharma Services supports customers with regulatory filings.
Aptar Pharma has consciously adopted the strategy of providing a wide range of support and services that customers need to advance their drug-device combination products through the regulatory process (Table 1).
| Ophthalmic Combination Device Support Documentation (related to Aptar Pharma device component) | Support from Aptar Pharma |
| Letter of Authorisation (LoA) | ✔ |
| Design Input (based on 21 CFR820.(c)) | ✔ |
| Design Output (based on 21 CFR820.30 (d)) | ✔ |
| Design Review (based on 21 CFR820.30 (e)) | ✔ |
| Design Verification (based on 21 CFR820.30 (f)) | ✔ |
| Design Validation (based on 21 CFR820.30 (g)) | Product Owner |
| Design Transfer (based on 21 CFR820.30 (h)) | ✔ |
| Design Changes (based on 21 CFR820.30 (i)) | ✔ |
| Design History File (based on 21 CFR820.30 (j)) | ✔ |
| Risk Management | ✔ |
| Supplemental (Human Factors etc.) | ✔ |
| Robustness Testing, Transport Studies | ✔ |
| Microbiology with Aptar Pharma Product and Support for Testing in Formulation | ✔ |
| Documentation of Sterilisation of Aptar Pharma Product | ✔ |
| Extractables Package for Aptar Pharma Product | ✔ |
| Additional Support Services with Customer Formulation, e.g. | |
| Device Formulation Acceptance Test Services | ✔ |
| Device Performance with Product Formulation | ✔ |
| Pre-Support for PAI or US FDA inspection at Manufacturer | ✔ |
| CMC Consulting Support | ✔ |
| EU General Safety and Performance Requirements Support | ✔ |
Table 1: Drug-device combination product documentation and service support considerations.
This includes topics such as design history files, design control documentation, verifications and custom performance evaluations of the devices, as well as the customer’s intended use and formulations. With decades of experience working hand in hand with customers, Aptar Pharma provides input on aspects such as device selection, human factors study design and materials that help to de-risk the product. The company not only designs and manufactures the devices but also offers integrated testing services, formulation development expertise and material compatibility analysis in its laboratories, carried out by its team of industry-leading experts. Aptar Pharma can look at physical drop characteristics and formulation parameters to allow the customer formulation to work with the selected device. In doing all of this, Aptar Pharma also provides the data and documentation for inclusion in – or in support of – regulatory filings. This is so that customers do not have to invest in developing the same specialised knowledge and expertise themselves.

Figure 5: Aptar Pharma’s broad expertise in analytical and regulatory services helps to advance drug-device combination products through the regulatory process.
Aptar Pharma is a full-service organisation, with services and partners that can support clinical and commercial manufacturing at virtually any scale (Figure 5). Moreover, Aptar Pharma continuously strives to develop and improve its analytical and regulatory support further, to deliver optimised, state-of-the-art procedures with patient safety in mind.
With increasing demand for PFMD eye-dropper technology, and more strict regulatory filing requirements for drug-device combination products, it is of critical importance to select a partner that not only provides a secure supply of high-quality and functional devices but can also provide the additional support, expertise and documentation on the device required to meet the filing and review requirements of regulatory authorities. Aptar Pharma continues to keep its eyes on a shared vision with its customers – that is “bringing new ophthalmic products to market that improve patient lives”.
To learn more about preservative free eye drops, visit: www.aptar.com/resources/an-eye-on-the-future-of-preservative-free-drops
REFERENCES
- “Ophthalmic Drugs Market Report to 2030”. Visiongain Report, Nov 26, 2020.
- Birkhoff M, “Delivering on theGrowing Need for Topical Preservative-Free Ophthalmic Treatments”. ONdrugDelivery, Issue 104 (Jan 2020), pp 8–12.
- Kahook M, “The Pros and Cons of Preservatives”. Review of Ophthalmology, Apr 15, 2015.
- Vaede D et al, “Preservatives in Eyedrops: Toward Awareness of their Toxicity”. Journal of Fr Ophtalmol, 2010, Vol 33(7), pp 505–524.
- Baudouin C et al, “Preservatives in Eyedrops: The Good, The Bad and The Ugly”. Prog Retin Eye Res, 2010, Vol 29, pp 312–334.
- Riau A et al, “Surface Immobilization of Nano-Silver on Polymeric Medical Devices to Prevent Bacterial Biofilm Formation”. Pathogens, 2019 Vol 8(3), p 93.
- Hasegawa A et al, “Microbiological Considerations for Ophthalmic Products: Sterility, Endotoxin Limits, and Preservatives”, Part II, pp 199–228 in Neervannan S, Kompella U (Eds), “Ophthalmic Product Development: From Bench to Bedside”, Advances in the Pharmaceutical Sciences, AAPS Springer, Book 37, 2021.
- “Combination Products Guidance Documents”. US FDA. Accessed Mar 4, 2022.
- “Guideline on the quality requirements for drug-device combinations”. EMA, May 2019.
- Bayarri L, “Drug-device combination products: regulatory landscape and market growth”. Drugs Today (Barc), 2015, Vol 51(8), pp 505–513.
- “MDR Article 117: Implications for Drug-device Combination Products”. Celegence. Company web page, accessed Mar 4, 2022.
- “Genus Medical Technologies LLC Versus Food and Drug Administration; Request for Information and Comments”. US FDA, Aug 2021.
- Losantos I, “Delivery to the eye: overcoming complex regulatory hurdles for combination products”. Cambridge Consultants Opinion, Jun 9, 2021.
- “Questions & Answers for applicants, marketing authorisation holders of medicinal products and notified bodies with respect to the implementation of the Medical Devices and In Vitro Diagnostic Medical Devices Regulations ((EU) 2017/745 and (EU) 2017/746)”. EMA, Jun 23, 2021.
- “Guideline on quality documentation for medicinal products when used with a medical device”. EMA, July 22, 2021.
- “Regulation (EU) 2017/745 of the European Parliament and of the council”. Apr 5, 2017.
- Marx D et al, “Microbial integrity test for preservative-free multidose eyedroppers or nasal spray pumps”. Pharm Ind, 2019, Vol 81(9), pp 1247–1252.