To Issue 145
Citation: Jones M, Veasey R, “Due Diligence – the Key to Success in Drug Delivery Device Selection”. ONdrugDelivery, Issue 145 (Apr 2023), pp 20–24.
Matt Jones and Rob Veasey consider the process of matching the drug with the right delivery device and look at the importance of due diligence when “licensing in” technology.
One of the first questions a pharmaceutical or biotech company should ask when bringing a new pulmonary or intranasal drug to market is whether they should “make” their own custom delivery device for it or “buy” a technology from a specialist delivery device developer.
“It is essential to go much further than simply comparing potential feature sets or reviewing key performance aspects on paper.”
Developing new drug delivery devices, or improving existing ones, is important to continue to improve the efficacy and reliability of treatments, as well as to increase the compliance of patients with their treatments. The regulatory pathways are set up to reflect this ethos of continual improvement – what was acceptable in the past may not be anymore. However, if devices are already available that are suitable for a new drug, “licensing in” device technology can be a preferable option. This is because, depending on the quality, maturity and suitability of the device and vendor, it can be quicker, lower risk and require less up-front investment, but this is a route that requires careful consideration to help ensure success.
While buying a device technology might theoretically offer a lower up-front investment and quicker route to market than developing one in-house, there are typically still significant sums of money involved, and choosing the wrong device can risk encountering major problems. There have been several instances where pharmaceutical companies have made a bad device choice and subsequently needed help to resolve fundamental design issues. In these cases, there have often been significant programme delays and serious financial implications arising from the need to make late-stage changes.
There is a clear lesson to take from this: when selecting a delivery device, it is essential to go much further than simply comparing potential feature sets or reviewing key performance aspects on paper. It is important to have a thorough and effective process and to consider engaging an independent specialist with the right expertise in device development, who can review and understand potential risks at a deep technical level and provide suitable advice.
“It is important for the device customer to determine the outline of a best practice development process, helping to establish the right expectations.”
THE RIGHT APPROACH TO DUE DILIGENCE
Because the reviewer needs to be able to evaluate complex technical issues, an effective approach to due diligence for device selection relies on the same skillset and experience as delivery device design and development. The key difference is that at least some of the device design data has already been generated.
At the outset, it is important for the device customer to determine the outline of a best practice development process, helping to establish the right expectations. This outline can then be populated with the product information, documentation and evidence available from the supplier, after which key gaps can be identified.
Alongside this development process framework, a robust, evidence-based set of requirements for the device is required. As far as possible, these should be determined independently of the capabilities of any candidate devices and be free from “outside influences”, such as marketing materials or industry hype. They should be based on a suitable understanding of factors, such as the intended dosing regimen(s), the specific needs of the target user population (dexterity issues in geriatric populations, for example), the potential future drug portfolio and pipeline (when evaluating a device platform) and issues with similar devices on market.
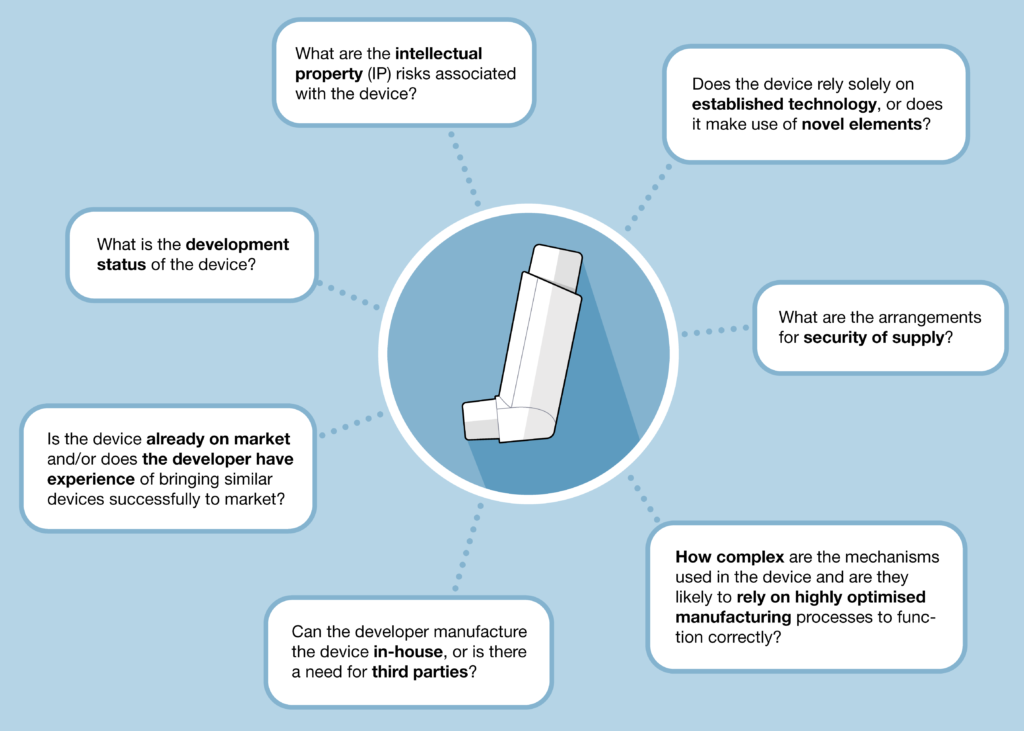
Figure 1: Some of the questions that should be asked in a candidate device assessment.
“Choosing a delivery device is an expensive and long-term commitment, so it is important to invest in the expertise that will ensure due diligence is properly performed.”
Next, a systematic risk assessment of candidate device performance and manufacturability should be undertaken. As an example, this assessment may ask questions such as those shown in Figure 1. The full list of questions for a given project will be much longer and should be tailored to the established requirements. Answering such questions will help generate an understanding of potential issues associated with a candidate device and allow for clearer judgements on the likelihood of potential delays or risks to the drug launch programme.
The key to success here is to have the support of a team with expertise and experience in drug delivery device development who can set out the process framework, establish the requirements and ask the right questions in a risk assessment to establish the suitability of a given candidate device. This expertise will need to encompass a range of disciplines, including usability, design, engineering, evaluation and, increasingly, software and electronics, all with specialist knowledge of the medical sector.
IS THIS REALLY NECESSARY?
It can be argued that, once a pharmaceutical company has chosen a device, the responsibility for ensuring its successful journey to market lies with the device developer. However, the reality is that no matter who bears responsibility on paper, once a pharmaceutical company has “bought in”, it is they who will primarily suffer the consequences of any failures or issues. These can be extremely serious – from lost investment and fines, through to reputational damage and launch delays (which, in themselves, could potentially lead to significant revenue loss or even losing the opportunity to participate in the market).
While it might seem appealing to rely on the expertise of the designers of the candidate devices, it is critical to delve deeper than the marketing pitch for each potential device and draw unbiased conclusions on its suitability for the intended application and any potential development risks it might pose. Choosing a delivery device is an expensive and long-term commitment, so it is important to invest in the expertise that will ensure due diligence is properly performed.
KEY FOCUS AREAS FOR EVALUATING DEVICES
During the device selection process, the specific areas of focus will depend on the requirements of the project. However, there are some common issues that regularly arise when licensing or purchasing “off-the-shelf” technology, which can be worth considering during initial evaluations.
Focus on Small-Scale Manufacturing Process
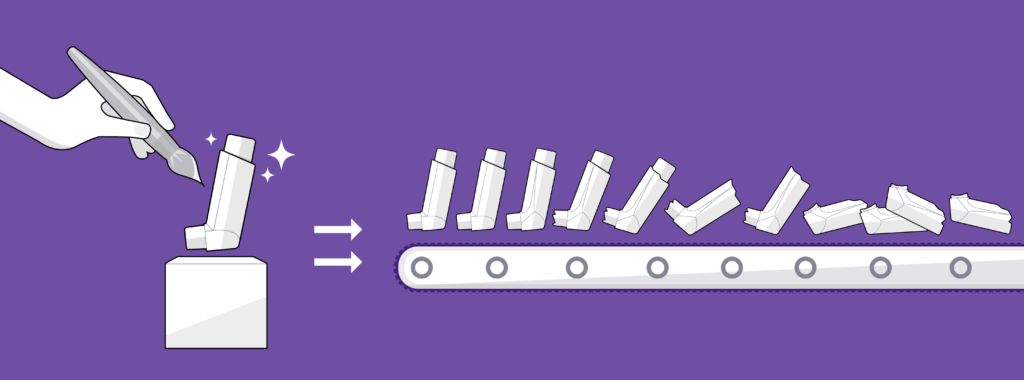
Figure 2: Focus exclusively on small-scale manufacturing processes can lead to problems.
Often, devices are designed with an exclusive focus on prototype manufacturing processes, the priority being to produce functional demonstrators and gain investment, with not enough consideration given to the process of manufacturing in larger volumes (Figure 2). This can manifest in a number of ways.
The first is that the component variation introduced when scaling-up to multicavity tooling can result in impaired device functionality or assembly issues. These issues can often be traced back to an incomplete or incorrect tolerance analyses. For example, some years ago, a pharmaceutical company requested a review of the design of a nasal spray device that was urgently needed for a clinical trial. The device was in multicavity production-ready tooling, but was found to perform erratically, with significant variation and degradation in spray performance over the intended operating life. The root cause was identified as a tolerance problem with the actuating mechanism. This had not been evident in earlier single cavity prototypes because the combination of component geometries had been favourable and had, therefore, been overlooked. This demonstrates the importance of scrutinising tolerance calculations for accuracy and completeness as part of the due diligence process.
The second is that the device components have not been designed with considerations for automated assembly. Component geometry may not incorporate the necessary features to enable efficient and effective feeding, gripping and manipulation with automated equipment. The interaction features may not have been designed with the speed and forces involved in automated assembly in mind, resulting in high scrap rates or unexpected damage during manufacture. For semi-manual lines, inadequate consideration may have been given to incorporating in-line checks and mitigating the possibility of human error.
The third is that a developer may not have given due consideration to control of the manufacturing process and supply chain. There should be an appropriate level of traceability for the raw materials (including pigments, paints and adhesives, for example) and the component manufacturing and assembly process parameters should be controlled and optimised. While an understanding of the level of scrutiny placed on these issues can be gleaned from the available documentation, manufacturing site visits can be one of several additional “tools” that can be highly beneficial for gaining further insight.
Usability and Design

Figure 3: Work done to explore usability and design risks should be examined.
It is important to establish whether the risks associated with user error, misuse and malfunction have been thoroughly considered, tested and transparently reported (Figure 3). To assess this, it is essential to start by determining whether the device developer has conducted a sufficiently detailed user risk assessment and component-level failure mode and effects (FMEA) analysis. These documents can also be a useful starting point for exploring the design history documentation of the device. For example, as part of a due diligence exercise for a major pharmaceutical company, they requested a review of the design and documentation for a dry powder inhaler that they were considering licensing. It was found that only a cursory risk analysis had been performed and a detailed FMEA was recommended. In undertaking this detailed analysis, the potential risk that a patient could receive a significant excess dose if the device were primed several times before inhaling was identified. This risk was confirmed by testing. This illustrates the value of early and detailed risk analysis, which considers not only correct use but also foreseeable misuse of the device.
Many modern drug delivery devices are designed as platforms that are intended to be adapted for use with a variety of medications and indications. This means that a version of the device may already have been launched, for example, by a competitor for a similar, or completely different, product. In these circumstances, it will very likely be important to consider the potential cost and time involved in customising the platform device to provide adequate differentiation, both from a risk (avoiding mix-up of medications) and brand (standing out against competitors) perspective. One not always obvious issue to consider here is whether or not you will get the level of service and timeliness you need to get your version of the product to the market as you need.
Intellectual Property
The drug delivery device intellectual property (IP) landscape is crowded. Therefore, it is important to understand this landscape for the device type being considered, the IP held by the device developer and their arguments as to why their device does not present a significant risk of infringement (Figure 4).

Figure 4: In depth focus on IP risk is critical for success.
As one of a number of assessment “tools”, it can be useful to challenge the device IP and its design by adopting a potential competitor’s mindset to consider how a candidate device could be argued to infringe, or why the developer’s IP might not be valid. These hypothetical counterarguments can then be considered against those of the candidate device developer for a more balanced assessment of potential risk.
One thing to be particularly aware of is that a track record of a device being on-market is not a guarantee of “freedom to operate”. It may only be when a device is combined with a particular drug that a competitor decides it is worth the effort and expense to launch an infringement challenge against it. There have been a number of recent high profile cases in the diabetes field in which exactly this scenario has occurred.

Figure 5: The quality and quantity of regulatory documentation available may not match expectations.
Regulatory Requirements
The level and quality of the documentation produced by the device developer may fall far short of what is necessary for successful regulatory submission in intended markets. It is therefore important to clarify exactly what documentation is required for submission and what will be provided by the device developer before committing to a candidate device (Figure 5). Having this understanding will be key to assessing what additional work will be needed for a successful submission. Particular attention should be paid to how any specific requirements of the drug in question may affect the submission file and what support the device developer is able to provide to fill any gaps.
It is often the case that important information is unavailable from the device developer, either because they simply do not have it or because they are restricting it to protect their IP – another situation that highlights the importance of independent expertise. To compensate for missing information, it is critical for the due diligence team to have a high level of technical understanding so that they can examine the situation and conduct assessments and tests of candidate devices.
CONCLUSION
Selecting a drug delivery device is not a simple process of browsing the available marketing materials for the device that appears to “fit the bill”. It requires a thorough and structured process to ensure that due diligence is undertaken to match a drug with the right delivery device and avoid costly mistakes. An effective due diligence process is fundamentally similar to developing a device from scratch, requiring many of the same skills, experience and expertise, which are highly specific and may not be readily available within a pharmaceutical company.
Ensuring that there is an effective approach to due diligence for device selection is easily overlooked but critical to the overall success of a drug development project. The benefits of getting it right can be enormous, and the consequences of getting it wrong can be dire. Partnering with independent experts with a proven record of drug delivery device design expertise can be the key to success, providing insight, de-risking a project and helping to pave the way to a smoother drug product launch and success on market.