
To Issue 173
Citation: Chow KT, “Interview: Patient Experience Versus Regulatory Compliance – Can Formulators Find a Middle Ground?”. ONdrugDelivery, Issue 173 (May/Jun 2025), pp 6–9.
Dr Keat Theng Chow outlines the essential themes on Roquette’s radar regarding patient-centric drug design, upcoming regulatory changes and how drug producers can navigate both spheres to the benefit of all.
Q How would you describe the current landscape when it comes to patient-centricity? Are we still in the era of top-down drug designs, or are patient needs exerting a greater influence?
“THE IMPORTANCE OF THIS APPROACH IS UNDERSCORED BY THE FACT THAT APPROXIMATELY 50% OF PEOPLE DO NOT TAKE MEDICATIONS AS PRESCRIBED, RESULTING IN SIGNIFICANT CONSEQUENCES FOR BOTH AN INDIVIDUAL’S HEALTH AND ENTIRE HEALTHCARE SYSTEMS.”
A The pharmaceutical industry is at a pivotal moment in its approach to drug design. Historically, the landscape has been dominated by a top-down methodology, where clinicians and drug developers determine what is best for patients in terms of treatment formats. This approach has contributed to significant medical advancements, but there is still opportunity for the needs and preferences of patients to be factored into the design process as early, and as regularly, as possible. The importance of this approach is underscored by the fact that approximately 50% of people do not take medications as prescribed,1 resulting in significant consequences for both an individual’s health and entire healthcare systems.2
Medical nonadherence is a multifaceted issue that most acutely affects people with chronic disease and older adults, though it can impact individuals of all ages and circumstances. Fortunately, the gradual shift away from the traditional one-size-fits-all approach to drug design towards one that prioritises individual patient preferences has led to the rise of improved coating and taste-masking solutions, controlled release and orally dispersible drugs, and – more recently – the incorporation of advanced technologies, such as 3D printing and artificial intelligence (AI), to support production.
Q Which specific dosage forms tend to be most popular among patients? Do these preferences differ depending on demographic factors such as age or region?
A The past decade has seen growing demand for more palatable and convenient alternatives to solid, swallowable pills, though the traditional tablet remains popular. In the pharmaceutical and nutraceutical sectors alike, chewable tablets and gummies have quickly climbed in popularity, particularly in the treatment of children, who typically find them more enjoyable and easier to swallow than standard pills. Similarly, liquids and syrups are often preferred by the parents of young children, as they offer greater flexibility in dosing and administration.
For older patients, focus has shifted to formats that address swallowing difficulties, such as orally disintegrating tablets (ODTs) and oral disintegrating films (ODFs). These dosage forms dissolve quickly in the mouth, eliminating the need for water and offering a convenient, easy-to-administer solution. Effervescent tablets and sachets are also widely used by older populations for their ease of use and, to a certain extent, separation from other drug formats that tend to trigger pill fatigue.
“TAKE CHEWABLE TABLETS, ODTs AND ODFs AS AN EXAMPLE; THE CHALLENGE AT THE HEART OF THESE DOSAGE FORMS IS THE TENSION BETWEEN LONG-TERM STABILITY AND OPTIMAL DISINTEGRATION.”
Q Can you provide specific examples of how Roquette’s offering helps pharma producers pursue more patient-centric drug design?
A Yes, certainly – APIs tend to be seen as the most important elements of pharmaceutical formulation but, when it comes to user-centric delivery, excipients are the true formulation heroes. Take chewable tablets, ODTs and ODFs as an example; the challenge at the heart of these dosage forms is the tension between long-term stability and optimal disintegration. To produce an end product capable of both, formulators need an excipient that combines excellent dispersibility with good mechanical strength, while simultaneously delivering a pleasant taste and texture. Co-processed mannitol-starches – such as our PEARLITOL® Flash solution – offer formulators excellent chemical inertness and a consistent, rapid disintegration performance, all with a pleasant flavour for maximum patient acceptance.
For manufacturers of more traditional pills, the core goals are to keep throughput speed and compression force high, and tablet sizes small to ensure maximum efficiency, quality and end-user convenience. Rapid processing rates and stronger compression forces can, however, increase the risk of capping or sticking – both of which lead to costly waste and a generally worsened patient experience.3
The solution for this is to select a direct-compression excipient with the optimal density and flow properties to maintain quality, even under the stress of high-pressure production. Our PEARLITOL® 200 GT mannitol was designed with these factors in mind, allowing for maximum compression force, tableting speed and API load, all without increased risk of capping or sticking. Indeed, a comparative study showed it was able to increase API load by over 100% while decreasing tablet weight by more than half.4 This potential to create smaller, harder, more stable tablets containing higher doses of APIs means that drug manufacturers can offer patients the simplified treatment plans they need to stay on track with essential medications.
Q Are expectations similarly in flux when it comes to regulatory standards? Are there any new developments that are having an especially significant impact on pharmaceutical producers right now?
A Yes – contrary to the perception of pharma regulations as slow moving, drug producers are often faced with near constant adaptations on a micro and macro scale. In recent months, volatile international trade regulations have hastened the introduction of measures, such as the EU’s proposed Critical Medicines Act and Health Technology Assessment Regulation, both of which aim to consolidate and accelerate the production of essential drugs within the EU. Regulators are simultaneously contending with emerging challenges, including the opportunities and risks posed by AI, and long-standing risks to patient health and safety that evolve in line with ongoing research.
One such example that is still unfolding is the conversation around nitrosamine impurities. Following more than five years of recommendations from authorities on both sides of the Atlantic, in January 2025, the US FDA published new guidelines for the establishment of acceptable intake limits for N-nitrosamine drug substance-related impurities (NDSRIs). In contrast to simpler, small-molecule nitrosamines, NDSRIs present complex and unique chemical structures, which makes the establishment of accurate acceptable limits particularly challenging. Formulators have therefore been forced to apply information from structurally similar compounds or conservative defaults to establish these safe limits, but this can lead to costly or even unnecessary production changes.
The FDA’s Carcinogenic Potency Categorization Approach (CPCA) proposes a new protocol that predicts the carcinogenic potency of an NDSRI by analysing its chemical makeup – whether known or merely theoretical. By combining scores awarded according to the structure of α-hydrogen atoms and the presence of activating or deactivating features, the CPCA assigns an NDSRI to a potency category, which then dictates its acceptable daily intake limit. This approach represents a notable breakthrough because it allows for a precise, risk-based assessment even in the absence of specific long-term carcinogenicity data for a particular NDSRI, making it an ideal case study for how regulatory updates don’t always mean extra work for pharmaceutical brands.
Q Patient needs are central, but they are not the only stakeholders involved in drug development – do the opinions of patentors and regulators conflict with that of patients? If so, how can this be overcome?
A Though they have a final goal in common, differences in experience and aim can sometimes result in contrasting perspectives on drug development between patients, patentors and regulators. There are, however, ways to overcome these differences. First, it’s important to recognise the various valid considerations that are most important to each group. For patients, convenience, effectiveness and accessibility are the primary concerns, while regulators must consider safety, efficacy and quality. Patent holders’ priorities, on the other hand, often centre on protecting intellectual property, securing returns on investment and navigating regulatory pathways to market.
Initiatives such as advisory boards or the inclusion of patient representatives in clinical trial design are effective methods to help bridge the gap between user expectations and other stakeholder priorities. Additionally, educational efforts at the community or frontline delivery level can help patients better understand potential regulatory constraints and the practicalities of drug product design. Through this two-way communication, all parties can align their goals and ensure that patients receive the safe, effective and accessible treatments they require.
Q How do you envision the future of patient-centric drug development? Are there any technologies or ingredients that could offer users more control or allow for closer collaboration between regulators, manufacturers and patients?
A We see the future of drug development as one where the best of advanced technologies and human ingenuity are harnessed to resolve the tension between patient experience and therapeutic efficacy. We’re already witnessing the roots of this in breakthroughs, such as AI-enhanced clinical studies, wearable health-tracking technology and smart pills, all of which could transform how drugs are designed, delivered and monitored in future.
The prospect of 3D-printed drugs moving from pipedream to tangible reality is an especially exciting step in the road to a tech-enabled, patient-centric future. Offering the potential to produce personalised medicines according to need, rather than the projected return on an industrial product run, 3D printing represents a whole new model for drug manufacturing with immense potential to enhance user experience.
“AS THE FIELD OF RESEARCH CONTINUES TO MATURE AND NEW, MORE REGULATOR FRIENDLY PROTOCOLS ARE LIKELY TO BEGIN TO EMERGE. WE THINK 3D PRINTING IS POISED TO BECOME THE NEXT LEAP FORWARD IN PATIENT-CENTRIC DRUG DELIVERY.”
One of the main hurdles that has prevented printed medicines from entering the mainstream up until this point is regulatory reticence. In 2015, the FDA granted approval for the first 3D-printed pill, SPRITAM® (levetiracetam, Aprecia Pharmaceuticals, Mason, OH, US) but, a decade on, it remains the only drug of its kind.5 The concerns here primarily relate to safety and consistency, with regulators questioning the fact that most 3D printing machines available to drug manufacturers were originally intended to produce plastics, rather than lifesaving pharmaceuticals.6 However, as the field of research continues to grow, more regulator-friendly protocols are likely to begin to emerge. We think 3D printing is poised to become the next leap forward in patient-centric drug delivery (Box 1).
BOX 1: STUDY SPOTLIGHT – EXPLORING APPLICATIONS OF PLANT-DERIVED POLYMERS IN FUSED DEPOSITION MODELLING OF ORAL PHARMACEUTICAL TABLETS
The material requirements for the most common form of pharmaceutical 3D printing, fused deposition modelling (FDM), have, in the past, been a significant source of regulatory scepticism. In short, the process requires the use of a filament polymer with the thermophysical ability to melt, pass through an extruder, fuse with previous layers and solidify quickly.7 Therefore, manufacturers often turn to synthetic oil-based materials, such as polyvinylpyrrolidone and polyvinyl alcohol, that, whilst effective, are not particularly popular with regulators or end users.8
Researchers at Roquette Health & Pharma Solutions recently set out to investigate an alternative approach to FDM production that allows for the use of widely known and trusted excipients, such as modified starches. The resultant study8 involved the testing of two model formulations:
- F1, which included the respiratory drug diprophylline as the API and pregelatinised hydroxypropyl pea starch as the polymer matrix excipient
- F2, which was a combination of pregelatinised potato starch and hydroxypropyl methylcellulose (HPMC K4M) alongside a range of APIs, namely diprophylline, theophylline anhydrous, caffeine anhydrous and indomethacin.
In both formulations, sorbitol and spray-dried mannitol were included as plasticisers, while stearic acid was used as a lubricant.

Figure 1: Photos of the tablets of F1 and F2, containing different APIs obtained by FDM.
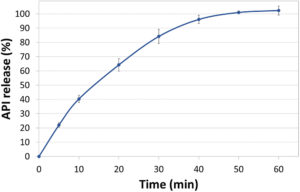
Figure 2: Dissolution profile of F1 diprophylline tablets tested using USP Apparatus 2 in SGF pH 1.2 media without enzymes. Error bars represent standard deviation (n=3).
The drug release kinetics of the resulting tablets (Figure 1) were evaluated in immediate and controlled release dissolution studies of 1- and 12-hour durations, some of the results of which can be seen in Figures 2 and 3. For the immediate release tests, tablets were dissolved in simulated gastric fluid (SGF) with a pH of 1.2, while the controlled-release varieties underwent a two-stage dissolution process in SGF at pH 1.2 for two hours, then in simulated intestinal fluid (SIF) at pH 6.8 for the remainder of the study.8
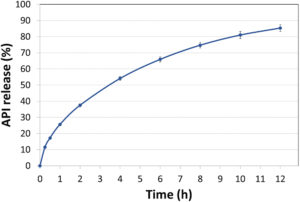
Figure 3: Dissolution profile of F2 diprophylline tablets tested using USP Apparatus 2 in SGF-SIF pH transition media without enzymes. Error bars represent standard deviation (n=3).
Results from the API release and later stability assessments showed that both base formulations containing hydroxypropyl starch and pre-gelatinised starch/HPMC (in combination with sorbitol and mannitol) were able to yield extrudable filaments with good printability, capable of achieving immediate and controlled API release for Biopharmaceutical Classification System Class 1 drugs.6 These findings point to a more regulatory-friendly approach to 3D-printed medications, which could in turn pave the way for a more patient-focused road ahead for drug manufacturing.8
Q Do you have any final thoughts on the intersection between patient experience and regulatory compliance?
A Whether drug manufacturer, safety inspector, patient advocate or excipients expert, the end goal is the same – better health and wellbeing for all. Ultimately, collaboration and empathy are the keys to achieving this aim, though advancing technology also has a significant role to play. These are the simple facts I see carrying the industry towards a future where regulation and user experience are two halves of the same whole.
REFERENCES
- Brown MT, Bussel JK, “Medication adherence: WHO cares?”. Mayo Clin Proc, 2011, Vol 86(4), pp 304–314.
- “€125 billion lost each year across Europe due to non-adherence to medication”. International Longevity Centre UK, Apr 2022.
- “Preventing Capping and other Tablet Defects”. Company Web Page, Natoli, accessed May 2025.
- Lefèvre P et al, “Optimizing API Load and Minimizing Tablet Weight Leveraging an Innovative DC Mannitol”. Presentation, 14th PBP World Meeting, Mar 2024, Vienna (Austria).
- Sultana N et al, “3D Printing in pharmaceutical manufacturing: Current status and future prospects”. Mater Today Commun, 2024, Vol 38, art 107987.
- Milliken RL et al, “Application of 3D printing in early phase development of pharmaceutical solid dosage forms”. Int J Pharm, 2024, vol 653, art 123902.
- H Peng et al, “3D printing processes in precise drug delivery for personalized medicine”. Biofabrication, 2024, Vol 16(3), art 032001.
- Chow KT et al, “Exploring Applications of Plant-Derived Polymers in Fused Deposition Modeling of Oral Pharmaceutical Tablets”. Presentation, AAPS 2023 PharmSci360, Oct 2023, Orlando (FL, US).