

To Issue 147
Citation: Burke J, “Tackling Design Challenges in Emergency-Use Autoinjectors.” ONdrugDelivery, Issue 147 (May 2023), pp 30–33.
John Burke considers the challenges that manufacturers need to overcome when designing emergency-use autoinjectors.
“Companies seeking to bring new emergency-use devices to market
are now faced with the challenge of demonstrating the 99.999% reliability required by the FDA, while also addressing known use- issues.”
It seems obvious that when developing a device to deliver a potentially life-saving medication, you want to ensure it is safe, reliable and easy to use. Achieving this with an emergency-use autoinjector poses significant challenges, in part due to the fact that they need to be used in highly stressful situations by a wide range of users. To date, several on-market devices have shown various use-related issues, while technical failures have resulted in a number of devices being recalled both in Europe and the US.
To counter this, the US FDA issued new guidance in 2020 outlining their expectations in terms of essential performance requirements and device reliability. While this has added clarity for what is required, the bar has been set high. Companies seeking to bring new emergency-use devices to market are now faced with the challenge of demonstrating the 99.999% reliability required by the FDA, while also addressing known use-issues. Achieving this requires a careful balance of user-centred design, regulatory strategy and design for reliability.
EMERGENCY-USE USER INTERFACES – WHAT ARE THE KNOWN ISSUES?
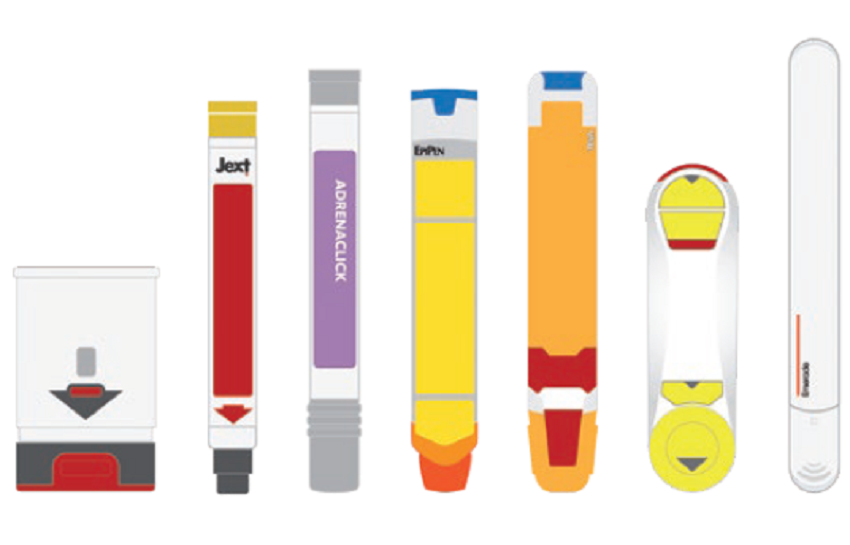
An essential aspect of emergency-use autoinjectors is ease of use. When looking at the devices currently available, a number of which are shown in Figure 1, there is a wide variety of different and potentially contradictory approaches to the user interface. It is therefore easy to see why confusion and user-error can occur. One study found that only 16% of adults who had been prescribed an adrenaline (epinephrine) autoinjector knew how to use the device correctly, including parents who might need to inject their child. This issue is not limited to a single device either. Another study in 2010 compared four devices – INT01, INT02, EpiPen® (Mylan, part of Viatris, PA, US) and TwinJect® (Verus Pharmaceuticals, CA, US) – and identified 13 different types of use error, including:
- Issues with the safety caps – removing in the wrong order, difficulty removing, or not removing at all
- Unintentional injection into the hand or digit
- Attempting to inject more than once
- Not holding for correct amount of time
- Attempting to disassemble the device
- Not injecting at all.

Figure 1: Currently available emergency-use autoinjectors pictured left to right: (Auvi-Q®, Jext®, Adrenaclick®, EpiPen®, Teva Generic®, Maverick®, Emerade®).
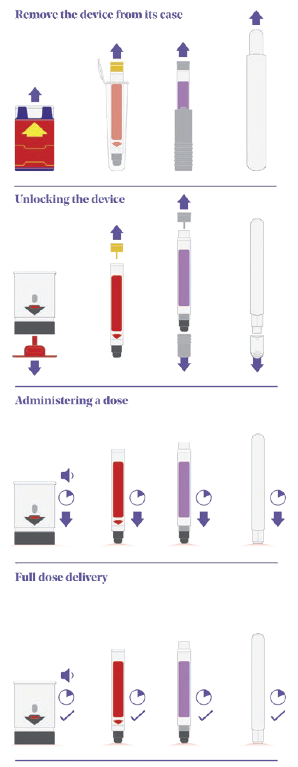
Figure 2: A comparison of emergency-use autoinjectors, pictured left to right (Auvi-Q®, Jext®, Adrenaclick®, Emerade®).
While the design of some of these devices has evolved since this 2010 study, there is still a variety of user interfaces among devices currently on the market.
SEQUENCE OF USE – A COMPARISON OF ON MARKET DEVICES
Figure 2 shows a comparison of four on-market emergency-use devices – Auvi-Q® (Kaléo), Jext® (ALK, Berkshire, UK), Adrenaclick® (Meridian Medical Technologies, MI, US) and Emerade® (Medeca Pharma, Uppsala, Sweden) – that clearly highlights some of the differences in their sequences of use and the potential root causes of user error:
- Remove the device from its protective case: Even with this simple step, several known use issues have occurred. During formative studies, users of the Auvi-Q® struggle to understand what to do with the case or lack the physical capability to remove the device from its tight-fitting sleeve. In other studies focused on different devices, there have been instances where users mistakenly believed the device was unlocked and ready to go once it had been removed from its protective case.
- Unlocking the device: The Auvi-Q® and Emerade® devices are both unlocked by pulling a safety feature or cap off the needle end of the device. Jext®, however, has a safety release that is pulled off the opposite end of the device, while Adrenaclick® requires the user to pull caps off both ends of the device (note the order is important here too – first the front and then the back).
- Administering a dose: All four devices actuate by firmly pushing the needle end against the skin to release the internal mechanism and start the injection. A common use issue that arises at this step is users holding the device in the wrong orientation. With more “traditional” devices, such as EpiPen® and Jext®, removing the safety release also leaves a round hole in the top of the device, which can result in accidental thumb injection. It should be noted that incorrect orientation is not limited to this type of device and has also been observed within studies evaluating devices with a more contemporary two-step interface as well. Various factors can impact this, including form-factor, any on-device instructions or cues, the use of colour and packaging and labelling.
- Full dose delivery: Once the device has been activated, the user needs to hold the device in place for between 3 and 10 seconds (depending on the device) before removing. Some devices have an indicator, but they are typically small and hard to see, while Emerade’s is under a peelable label. The Auvi-Q® device talks the user through the process and provides a clear audible countdown, which can be helpful.
When ease of use is paramount, this variety in user-interfaces places additional demands on the user, so it is hardly surprising to see use errors. A standardised sequence of use could help reduce the instances of use-error; however, it is important to note that this approach can introduce its own challenges as well.
Can a Contemporary “Two-Step” Approach Apply to Emergency-Use Autoinjectors?
In the last five years, a simple “two-step” user interface has become popular among autoinjectors used to treat chronic conditions such as rheumatoid arthritis, Crohn’s disease and psoriasis. There are, of course, outliers, but the benefits of a single, intuitive interface are clear. The FDA and other regulatory bodies have also become familiar with the approach and endorse the idea of standardisation.
“Generic device manufacturers must carefully balance the challenges of ensuring enough similarity to the existing device, while simultaneously tackling the known use issues that may accompany it.”
It seems logical therefore that, if you were developing a new emergency-use autoinjector, you would shift to this more contemporary two-step approach. Not only is it proven in other applications and fast becoming industry standard, it would help remove some of the known use-errors associated with traditional devices.
The challenge is that millions of users have already been trained and are familiar with existing emergency-use devices. Any changes made to existing devices presents a potential risk. This does not mean it is the wrong thing to do, but every effort needs to be taken to reduce the risk of misuse due to established mental models or previous device experience.
DESIGNING FOR USABILITY
Regulatory Strategy
The regulatory strategy a device manufacturer adopts will have an impact on the user interface of their product. This is particularly apparent for generic device design. Currently, many emergency-use devices contain drugs that have already been approved for use in the US. Section 505(j) of the Federal Food, Drug, and Cosmetic Act enables companies to demonstrate “sameness” to a reference listed drug (RLD) and leverage some existing safety and efficacy data, helping to save on clinical trials or other expensive studies. For this to apply, however, the drug product must be “therapeutically equivalent” and the accompanying device similar to the RLD, so that prescribing doctors and users can be confident in its use.
In addition to 505(j), the FDA has issued specific guidance on the design of generic adrenaline autoinjectors. The guidance does not state that the design should be identical, however the FDA is clearly conscious that manufacturers are not designing in a vacuum and that users may have experience or existing mental models around existing (potentially flawed) devices. What the FDA will not accept is manufacturers not addressing areas of the device that are known to cause confusion or issues – they will want to see evidence that these risks have been effectively mitigated.
In 2018, the FDA approved the first generic adrenaline injector from Teva Pharmaceuticals (Tel Aviv, Israel) as an ANDA under 505(j). Notably, the device has several key differences in the user interface to the RLD. For example, the sequence of use has changed from two steps to three with the introduction of a twist of cap covering the needle end, while the blue safety release differs somewhat from the RLD as it peels off from one side rather than pulling straight up.
Generic device manufacturers must carefully balance the challenges of ensuring enough similarity to the existing device, while simultaneously tackling the known use issues that may accompany it.
The Importance of Human Factors Engineering
Regardless of the regulatory strategy chosen, it is essential to have an effective human factors engineering (HFE) programme that runs in parallel with the design and engineering activities in accordance with IEC 62366-1. By having HFE as an integral part of the process from the outset, it is possible to optimise the device design, identify flaws and mitigate use errors. Every touchpoint is an opportunity to improve the usability of the device and its full ecosystem, from the physical device design, labelling and instructional information through to the packaging and more.
By including HFE through the entire development process, an HFE summary report can be submitted to the regulators that shows how the design has been optimised to minimise the potential for use error.
User capability studies can inform the specification limits, which, in turn, form the basis of essential performance requirements (e.g. the force to remove the cap or actuate the device). These need to be very carefully considered, as they form the target the manufacturer will be held to from a reliability standpoint. If the limit is too high, there is a risk that users may not be physically capable of performing the task. If the range is too narrow, it may be impossible to demonstrate the required level of reliability consistently.
Designing for Reliability – FDA Draft Guidance
Once the user interface has been considered and optimised, the next challenge is reliability. The FDA draft guidance on the reliability of emergency-use injectors, published in April 2020, has brought some clarity about what the FDA expects. The guidance describes the application of a scored fault tree analysis (FTA), alongside traditional development activities and approaches for achieving a reliability of 99.999% with a 95% level of confidence for the device. FTA is a well-established risk analysis and trouble-shooting tool that uses a top-down approach, starting with the main fault/effect and working down to potential root causes. Typically, an initial FTA is developed early in the development process, after which predicted probability can be applied based on simulation, design analysis, initial testing and informed manufacturing assumptions.
The required level of reliability to be demonstrated is understandably high. Device manufacturers should not underestimate the challenge of achieving and demonstrating that their device meets these requirements.
THE SUSTAINABILITY FACTOR
So far, this article has focused on two main areas: usability and reliability. There is, however, another important design consideration: sustainability. In practice, many emergency-use injectors are never used, with the majority expiring and being disposed of before they are required. It therefore makes sense to consider the environmental impact of decisions around the design, assembly and supply of components. From experience in conducting lifecycle analysis – a tool used to determine the carbon footprint of component manufacture and transportation – the greatest impact on device development is likely to come from:
- Supply change management, especially air travel
- Cold chain storage
- Device/packaging size
- Device architecture
- Drive (spring vs gas)
- Integration of electronics
- Shelf life
- Possibly shifting from glass primary packaging to copolymer or cyclic-olefin polymer.
These are all important considerations that should be factored in alongside designing for usability and reliability.
SUMMARY
Developing an emergency-use autoinjector is particularly challenging because of several issues:
- Usability – These products need to be used safely and effectively, every time; however, the usability of some of the devices currently available leaves a lot to be desired and known use errors persist. Adding to this is the challenge of shifting away from these potentially flawed interfaces due to existing mental models and the prior experience and training of users. To resolve this there is a clear need for a rigorous, effective HFE programme.
- Reliability – In terms of reliability and achieving “five nines”, while the FDA draft guidance provides clarity, the bar has been set very high. Conforming to this will significantly impact and shape the development of emergency-use devices.
- Sustainability – Finally, while it is critical not to compromise on the two key drivers above, it is also important to consider how the environmental impact of these single-use devices can be reduced, given their relatively short life and the number that end up in landfill.
Moving forwards, device manufacturers must balance each of these factors carefully to develop devices that are safe, effective and reliable, while minimising their carbon footprint.
